

A systematic bioinformatics approach for large-scale identification and characterization of host-pathogen shared sequences
- PMID: 34583643
- PMCID: PMC8477458
- DOI: 10.1186/s12864-021-07657-4
A systematic bioinformatics approach for large-scale identification and characterization of host-pathogen shared sequences
Abstract
Background: Biology has entered the era of big data with the advent of high-throughput omics technologies. Biological databases provide public access to petabytes of data and information facilitating knowledge discovery. Over the years, sequence data of pathogens has seen a large increase in the number of records, given the relatively small genome size and their important role as infectious and symbiotic agents. Humans are host to numerous pathogenic diseases, such as that by viruses, many of which are responsible for high mortality and morbidity. The interaction between pathogens and humans over the evolutionary history has resulted in sharing of sequences, with important biological and evolutionary implications.
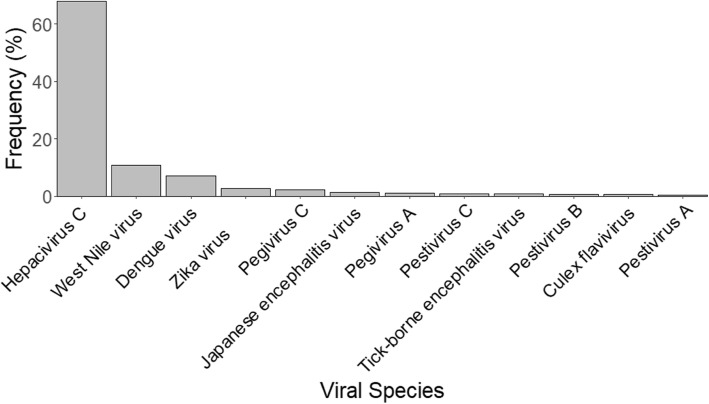
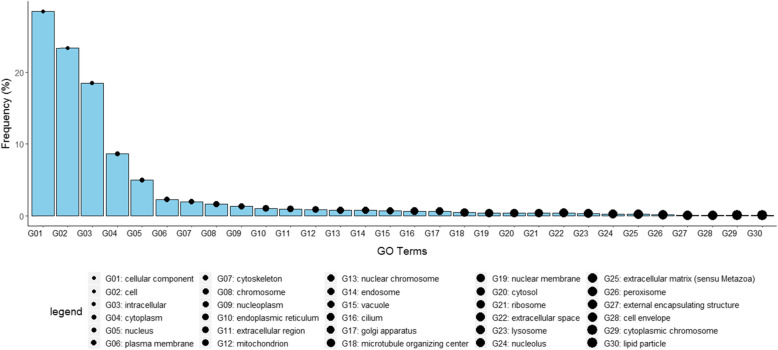
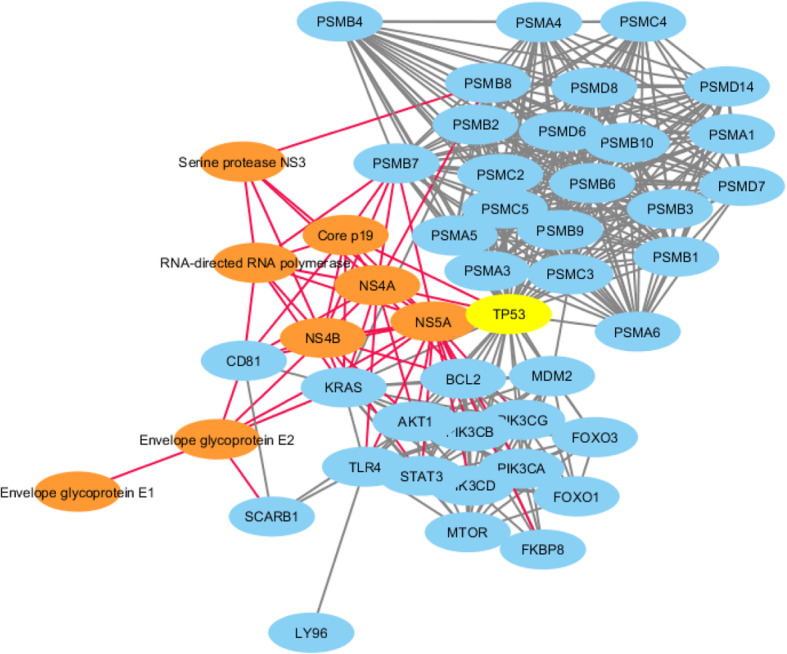
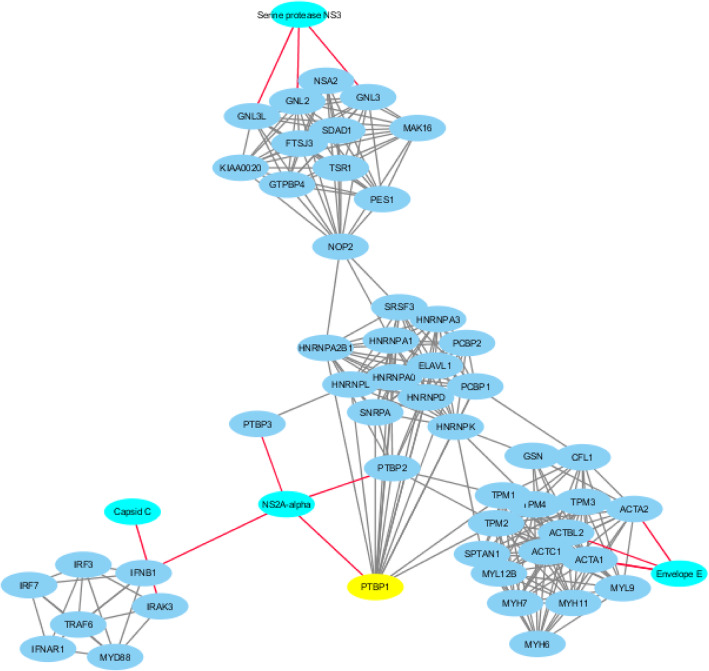
Results: This study describes a large-scale, systematic bioinformatics approach for identification and characterization of shared sequences between the host and pathogen. An application of the approach is demonstrated through identification and characterization of the Flaviviridae-human share-ome. A total of 2430 nonamers represented the Flaviviridae-human share-ome with 100% identity. Although the share-ome represented a small fraction of the repertoire of Flaviviridae (~ 0.12%) and human (~ 0.013%) non-redundant nonamers, the 2430 shared nonamers mapped to 16,946 Flaviviridae and 7506 human non-redundant protein sequences. The shared nonamer sequences mapped to 125 species of Flaviviridae, including several with unclassified genus. The majority (~ 68%) of the shared sequences mapped to Hepacivirus C species; West Nile, dengue and Zika viruses of the Flavivirus genus accounted for ~ 11%, ~ 7%, and ~ 3%, respectively, of the Flaviviridae protein sequences (16,946) mapped by the share-ome. Further characterization of the share-ome provided important structural-functional insights to Flaviviridae-human interactions.
Conclusion: Mapping of the host-pathogen share-ome has important implications for the design of vaccines and drugs, diagnostics, disease surveillance and the discovery of unknown, potential host-pathogen interactions. The generic workflow presented herein is potentially applicable to a variety of pathogens, such as of viral, bacterial or parasitic origin.
Keywords: Bioinformatics; Cross-reactivity; Crossreactome; Dengue virus; Flaviviridae; Flavivirus; Hepacivirus; Hepatitis C virus; Host-pathogen; Large-scale; Methodology; Pegivirus; Peptide overlap; Peptide sharing; Pestivirus; Share-ome; Shared sequences; West Nile virus; and Molecular mimicry..
© 2021. The Author(s).
Conflict of interest statement
We the authors declare that we have no competing interests in the research.
Figures



References
-
- Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, Liu M, Kumar S, Zaremba S, Gu Z, Zhou L, Larson CN, Dietrich J, Klem EB, Scheuermann RH. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40(D1):593–598. doi: 10.1093/nar/gkr859. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
