

Targeting mutant p53 for cancer therapy: direct and indirect strategies
- PMID: 34583722
- PMCID: PMC8480024
- DOI: 10.1186/s13045-021-01169-0
Targeting mutant p53 for cancer therapy: direct and indirect strategies
Abstract
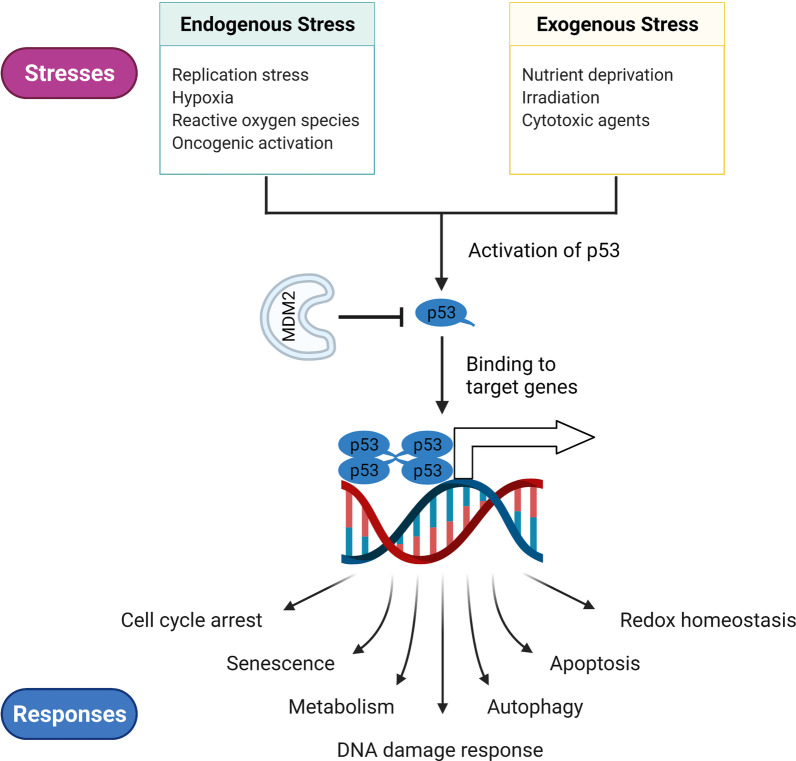
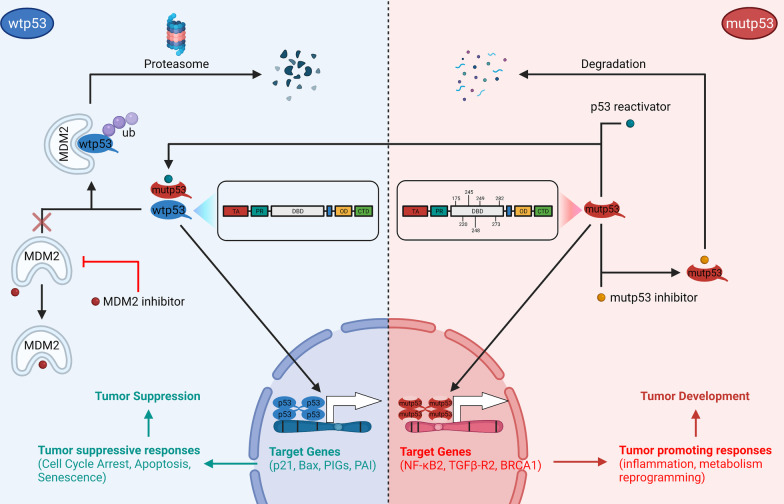
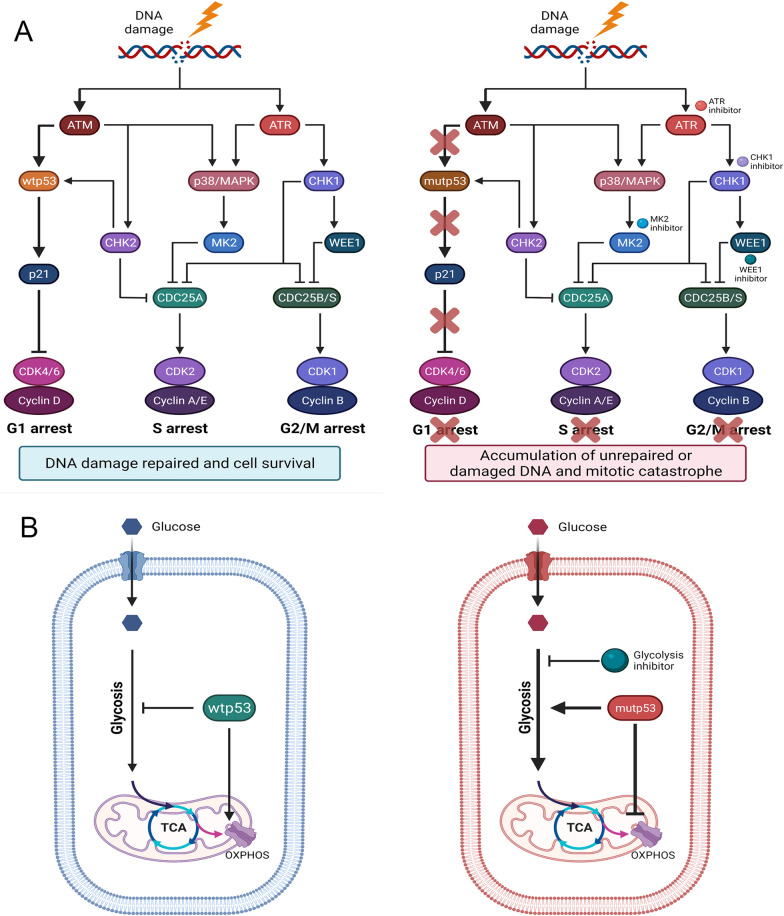
TP53 is a critical tumor-suppressor gene that is mutated in more than half of all human cancers. Mutations in TP53 not only impair its antitumor activity, but also confer mutant p53 protein oncogenic properties. The p53-targeted therapy approach began with the identification of compounds capable of restoring/reactivating wild-type p53 functions or eliminating mutant p53. Treatments that directly target mutant p53 are extremely structure and drug-species-dependent. Due to the mutation of wild-type p53, multiple survival pathways that are normally maintained by wild-type p53 are disrupted, necessitating the activation of compensatory genes or pathways to promote cancer cell survival. Additionally, because the oncogenic functions of mutant p53 contribute to cancer proliferation and metastasis, targeting the signaling pathways altered by p53 mutation appears to be an attractive strategy. Synthetic lethality implies that while disruption of either gene alone is permissible among two genes with synthetic lethal interactions, complete disruption of both genes results in cell death. Thus, rather than directly targeting p53, exploiting mutant p53 synthetic lethal genes may provide additional therapeutic benefits. Additionally, research progress on the functions of noncoding RNAs has made it clear that disrupting noncoding RNA networks has a favorable antitumor effect, supporting the hypothesis that targeting noncoding RNAs may have potential synthetic lethal effects in cancers with p53 mutations. The purpose of this review is to discuss treatments for cancers with mutant p53 that focus on directly targeting mutant p53, restoring wild-type functions, and exploiting synthetic lethal interactions with mutant p53. Additionally, the possibility of noncoding RNAs acting as synthetic lethal targets for mutant p53 will be discussed.
Keywords: Cancer therapy; MDM2 inhibitor; Noncoding RNA; Synthetic lethality; p53; p53 restoration.
© 2021. The Author(s).
Conflict of interest statement
All the authors declare no conflict of interests.
Figures
Similar articles
-
New therapeutic strategies to treat human cancers expressing mutant p53 proteins.J Exp Clin Cancer Res. 2018 Feb 15;37(1):30. doi: 10.1186/s13046-018-0705-7. J Exp Clin Cancer Res. 2018. PMID: 29448954 Free PMC article. Review.
-
Molecularly targeted therapies for p53-mutant cancers.Cell Mol Life Sci. 2017 Nov;74(22):4171-4187. doi: 10.1007/s00018-017-2575-0. Epub 2017 Jun 22. Cell Mol Life Sci. 2017. PMID: 28643165 Free PMC article. Review.
-
[Synthetic lethal genes to mutant p53].Yi Chuan. 2015 Apr;37(4):321-326. doi: 10.16288/j.yczz.14-277. Yi Chuan. 2015. PMID: 25881697 Review. Chinese.
-
Reactivation of mutant p53 by capsaicin, the major constituent of peppers.J Exp Clin Cancer Res. 2016 Sep 6;35(1):136. doi: 10.1186/s13046-016-0417-9. J Exp Clin Cancer Res. 2016. PMID: 27599722 Free PMC article.
-
Unraveling the Role of TP53 in Colorectal Cancer Therapy: From Wild-Type Regulation to Mutant.Front Biosci (Landmark Ed). 2024 Jul 25;29(7):272. doi: 10.31083/j.fbl2907272. Front Biosci (Landmark Ed). 2024. PMID: 39082342 Review.
Cited by
-
Spatial immunogenomic patterns associated with lymph node metastasis in lung adenocarcinoma.Exp Hematol Oncol. 2024 Oct 28;13(1):106. doi: 10.1186/s40164-024-00574-8. Exp Hematol Oncol. 2024. PMID: 39468696 Free PMC article.
-
Exploring the association between cancer and cognitive impairment in the Australian Imaging Biomarkers and Lifestyle (AIBL) study.Sci Rep. 2024 Feb 22;14(1):4364. doi: 10.1038/s41598-024-54875-3. Sci Rep. 2024. PMID: 38388558 Free PMC article.
-
Cell Death Pathway Regulation by Functional Nanomedicines for Robust Antitumor Immunity.Adv Sci (Weinh). 2024 Jan;11(3):e2306580. doi: 10.1002/advs.202306580. Epub 2023 Nov 20. Adv Sci (Weinh). 2024. PMID: 37984863 Free PMC article. Review.
-
Case Report: A Novel Pathomechanism in PEComa by the Loss of Heterozygosity of TP53.Front Oncol. 2022 Mar 28;12:849004. doi: 10.3389/fonc.2022.849004. eCollection 2022. Front Oncol. 2022. PMID: 35419288 Free PMC article.
-
Multifunctional nanoparticle for cancer therapy.MedComm (2020). 2023 Jan 11;4(1):e187. doi: 10.1002/mco2.187. eCollection 2023 Feb. MedComm (2020). 2023. PMID: 36654533 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
