

Interplay of m6 A and histone modifications contributes to temozolomide resistance in glioblastoma
- PMID: 34586728
- PMCID: PMC8441140
- DOI: 10.1002/ctm2.553
Interplay of m6 A and histone modifications contributes to temozolomide resistance in glioblastoma
Abstract
Background: Despite the development of new treatment protocols for glioblastoma (GBM), temozolomide (TMZ) resistance remains a primary hindrance. Previous studies, including our study, have shown that aberrant N6-methyladenosine (m6 A) modification is implicated in GBM pathobiology. However, the roles and precise mechanisms of m6 A modification in the regulation of TMZ resistance in GBM remain unclear.
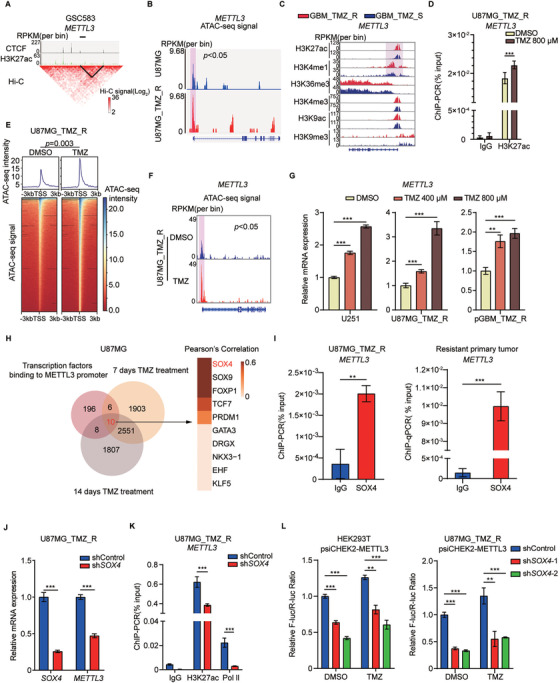
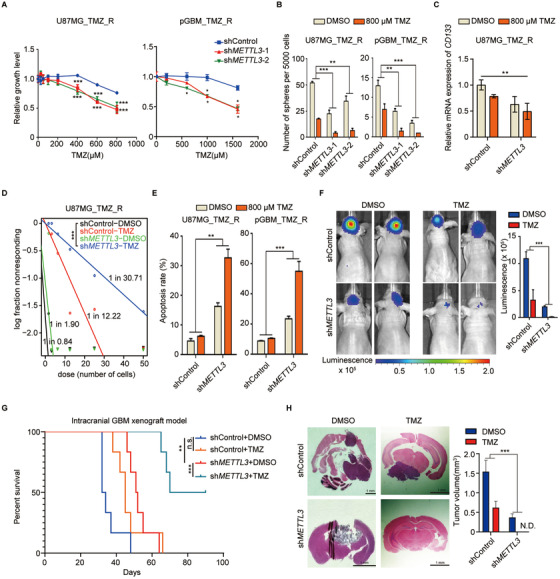
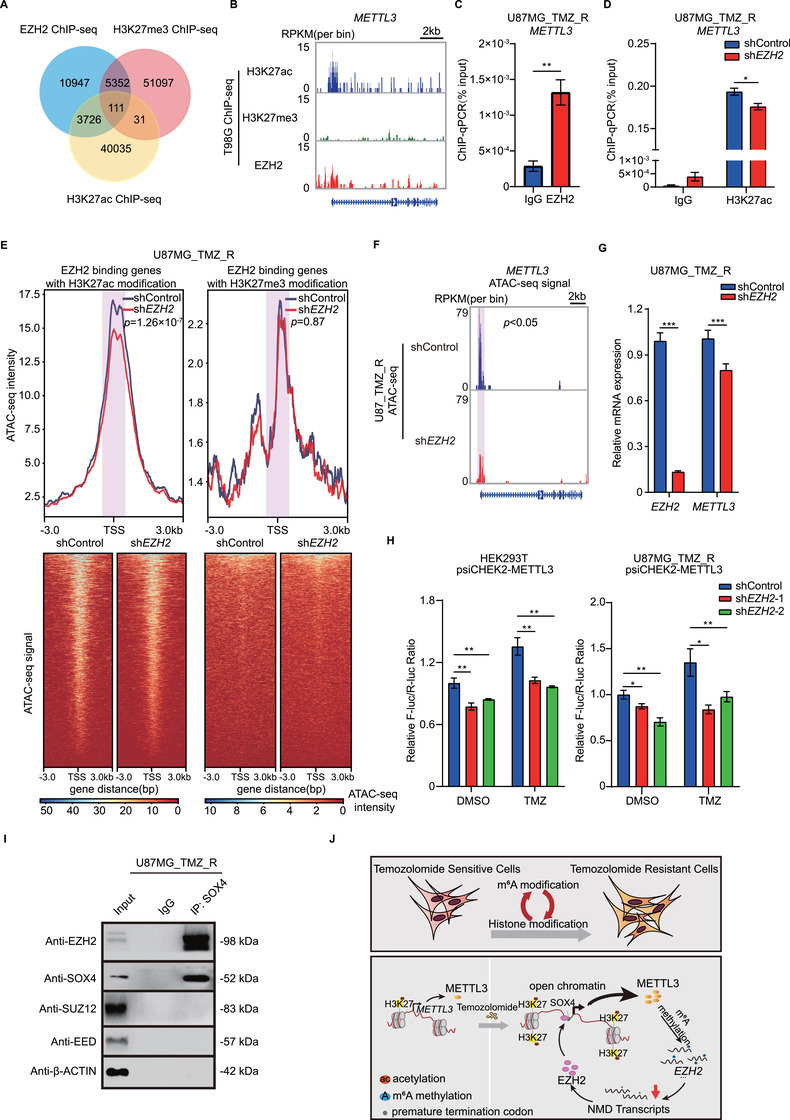
Methods: m6 A individual-nucleotide-resolution cross-linking and immunoprecipitation sequencing (miCLIP-seq) was performed to identify m6 A modification of transcripts in TMZ-resistant and -sensitive tumors. To explore the role of METTL3 in TMZ resistance, TMZ-resistant GBM cells were transfected with METTL3 shRNA or overexpression lentivirus and then assessed by cell viability, tumor sphere formation, and apoptosis assays. An intracranial GBM xenograft model was developed to verify the effect of METTL3 depletion during TMZ treatment in vivo. ATAC-seq, ChIP-qPCR, and dual-luciferase reporter assays were carried out to verify the role of SOX4/EZH2 in the modulation of METTL3 expression upon TMZ treatment.
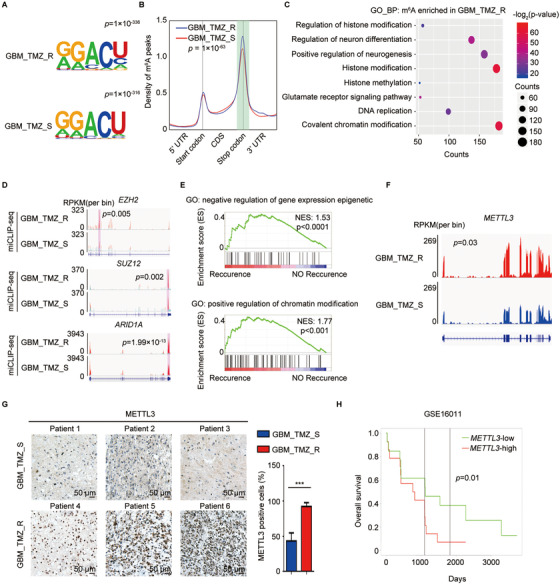
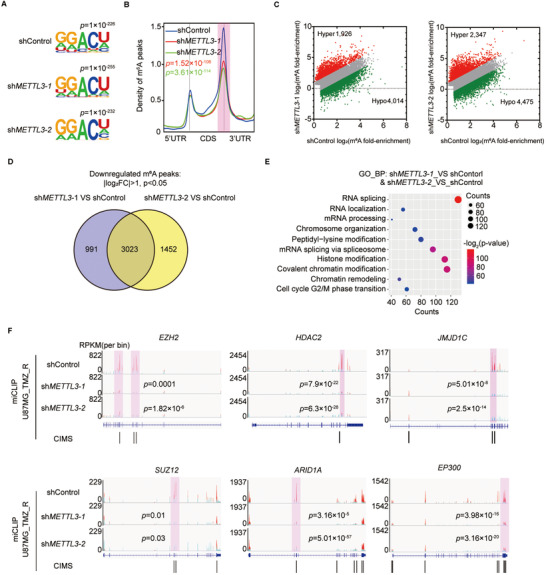
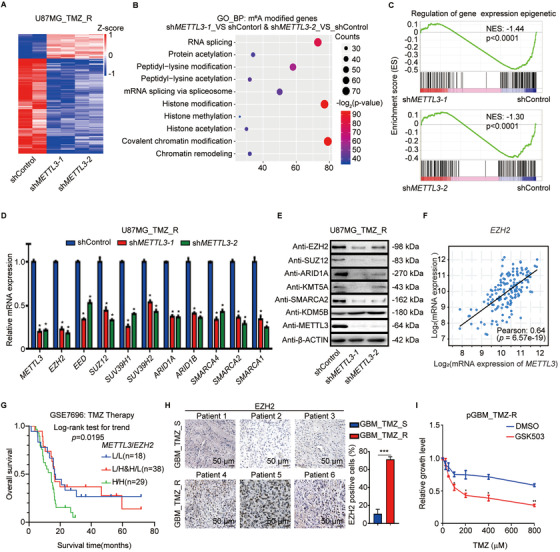
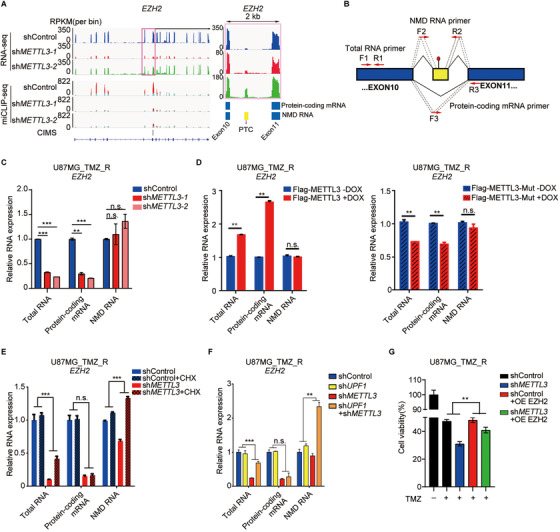
Results: We demonstrated that TMZ treatment upregulated the expression of the m6 A methyltransferase METTL3, thereby increasing m6 A modification of histone modification-related gene transcripts. METTL3 is required to maintain the features of GBM stem cells. When combined with TMZ, METTL3 silencing suppressed orthotopic TMZ-resistant xenograft growth in a cooperative manner. Mechanistically, TMZ induced a SOX4-mediated increase in chromatin accessibility at the METTL3 locus by promoting H3K27ac levels and recruiting RNA polymerase II. Moreover, METTL3 depletion affected the deposition of m6 A on histone modification-related gene transcripts, such as EZH2, leading to nonsense-mediated mRNA decay. We revealed an important role of EZH2 in the regulation of METTL3 expression, which was via an H3K27me3 modification-independent manner.
Conclusions: Our findings uncover the fundamental mechanisms underlying the interplay of m6 A RNA modification and histone modification in TMZ resistance and emphasize the therapeutic potential of targeting the SOX4/EZH2/METTL3 axis in the treatment of TMZ-resistant GBM.
Keywords: METTL3; TMZ resistance; glioblastoma; histone modifications; m6A.
© 2021 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
No potential conflict of interest to disclose.
Figures
References
-
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492‐507. - PubMed
-
- Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G. High‐grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2014;25(3):iii93‐101. - PubMed
-
- Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4(4):296‐307. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources