

A more accurate method for colocalisation analysis allowing for multiple causal variants
- PMID: 34587156
- PMCID: PMC8504726
- DOI: 10.1371/journal.pgen.1009440
A more accurate method for colocalisation analysis allowing for multiple causal variants
Abstract
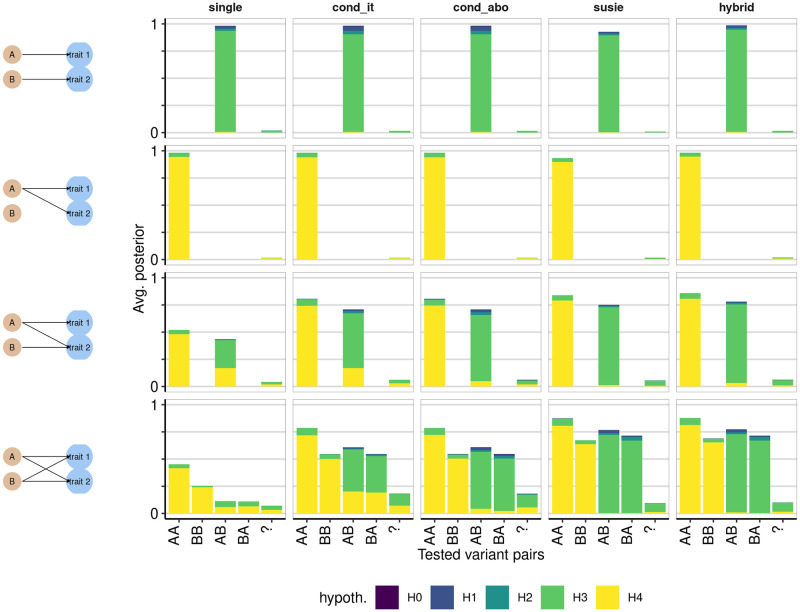
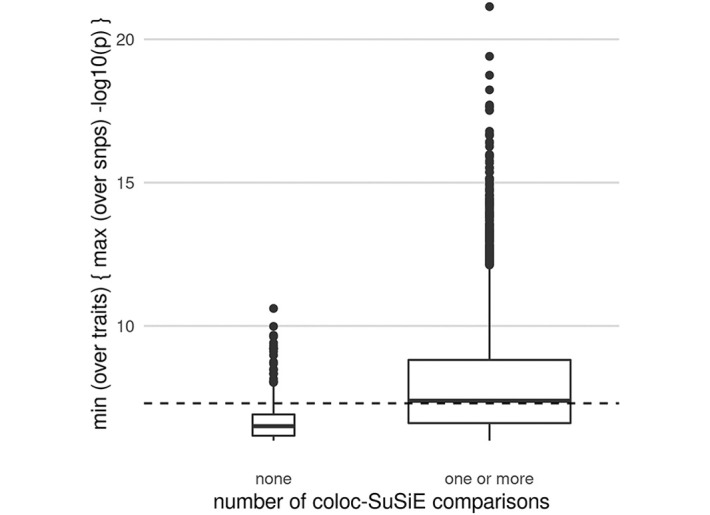
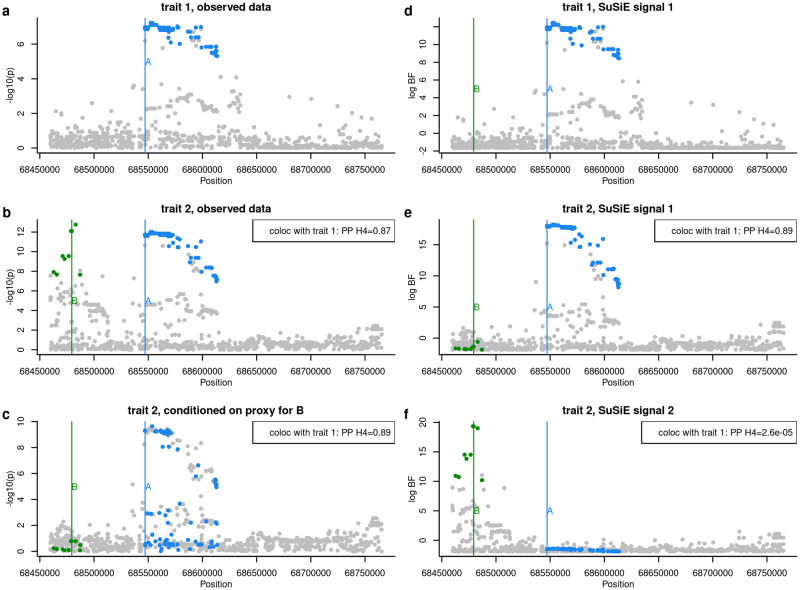
In genome-wide association studies (GWAS) it is now common to search for, and find, multiple causal variants located in close proximity. It has also become standard to ask whether different traits share the same causal variants, but one of the popular methods to answer this question, coloc, makes the simplifying assumption that only a single causal variant exists for any given trait in any genomic region. Here, we examine the potential of the recently proposed Sum of Single Effects (SuSiE) regression framework, which can be used for fine-mapping genetic signals, for use with coloc. SuSiE is a novel approach that allows evidence for association at multiple causal variants to be evaluated simultaneously, whilst separating the statistical support for each variant conditional on the causal signal being considered. We show this results in more accurate coloc inference than other proposals to adapt coloc for multiple causal variants based on conditioning. We therefore recommend that coloc be used in combination with SuSiE to optimise accuracy of colocalisation analyses when multiple causal variants exist.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLOS Genetics. 2014. May;10(5):e1004383. Available from: http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.10.... - PMC - PubMed
-
- Wallace C. Eliciting Priors and Relaxing the Single Causal Variant Assumption in Colocalisation Analyses. PLOS Genetics. 2020. Apr;16(4):e1008720. Available from: https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1.... - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
