

Blood and immune development in human fetal bone marrow and Down syndrome
- PMID: 34588693
- PMCID: PMC7612688
- DOI: 10.1038/s41586-021-03929-x
Blood and immune development in human fetal bone marrow and Down syndrome
Abstract
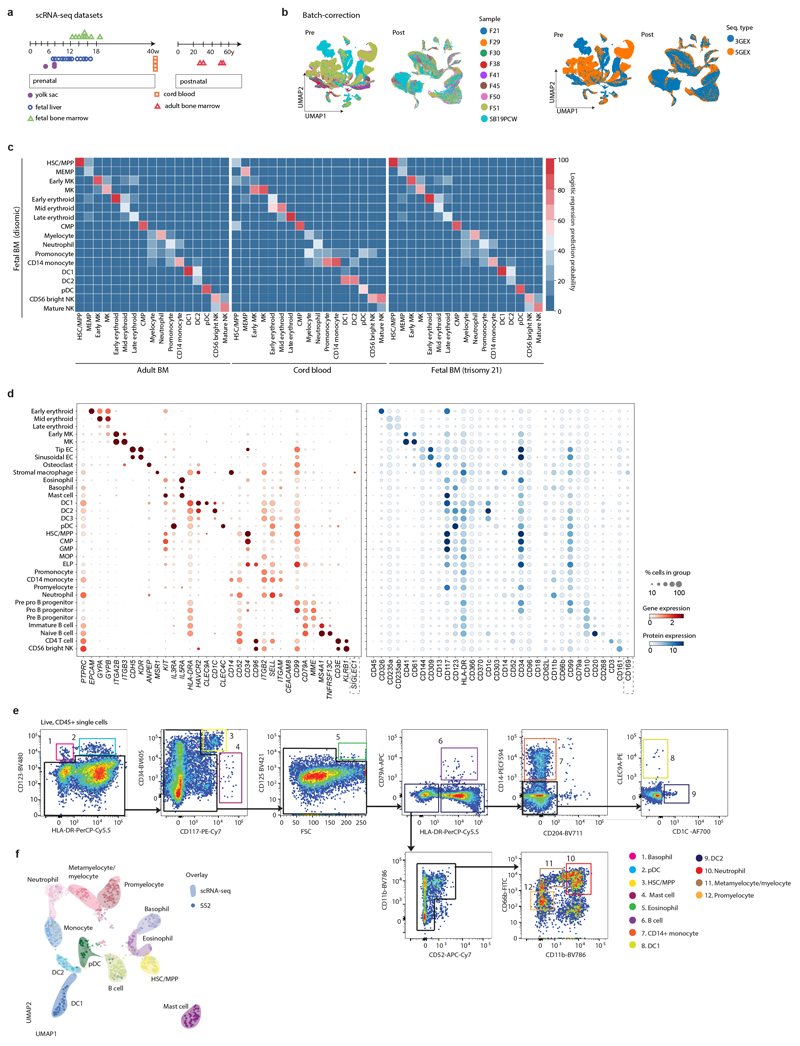
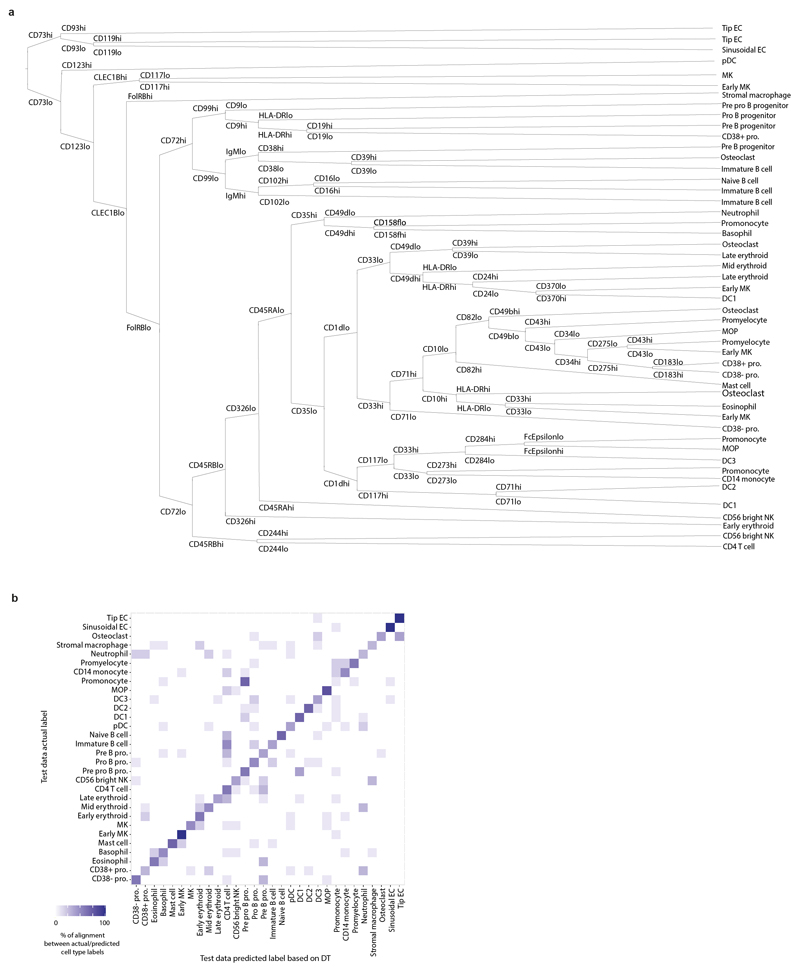
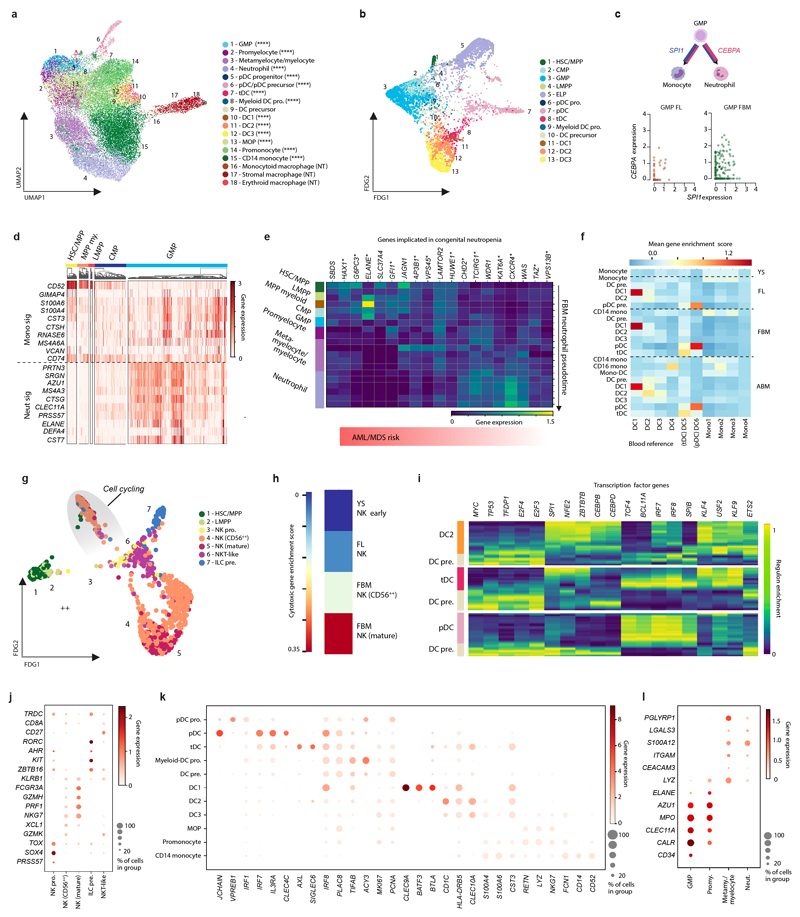
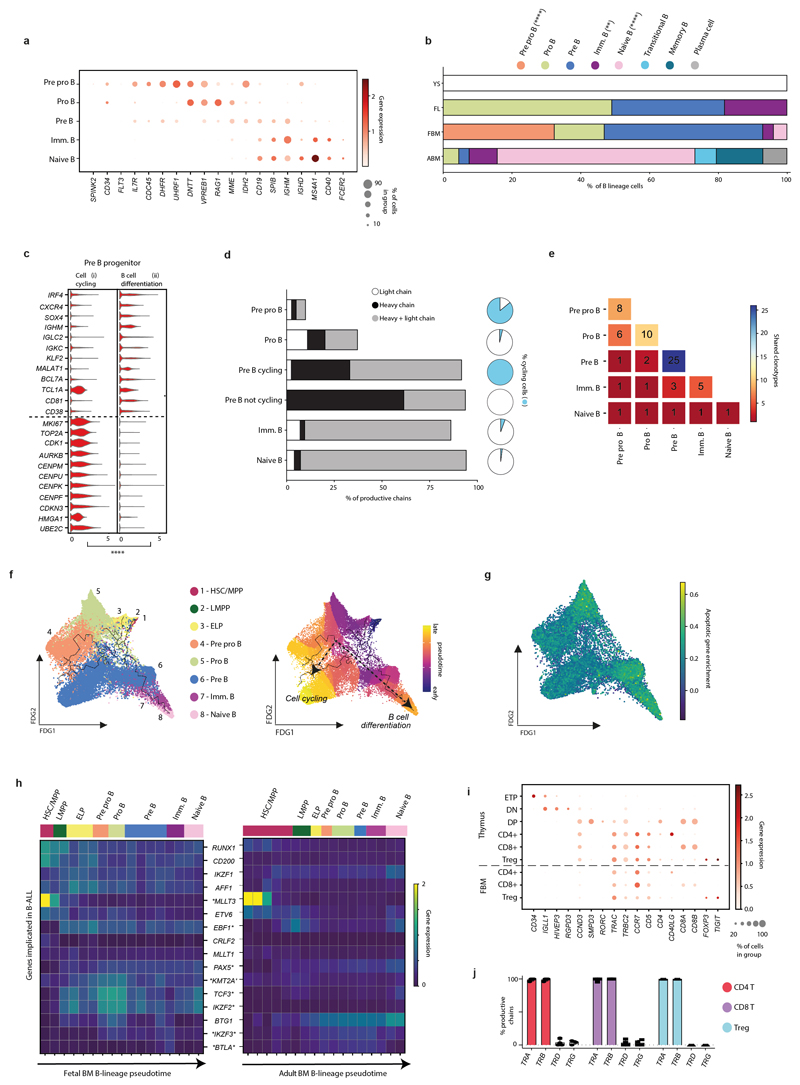
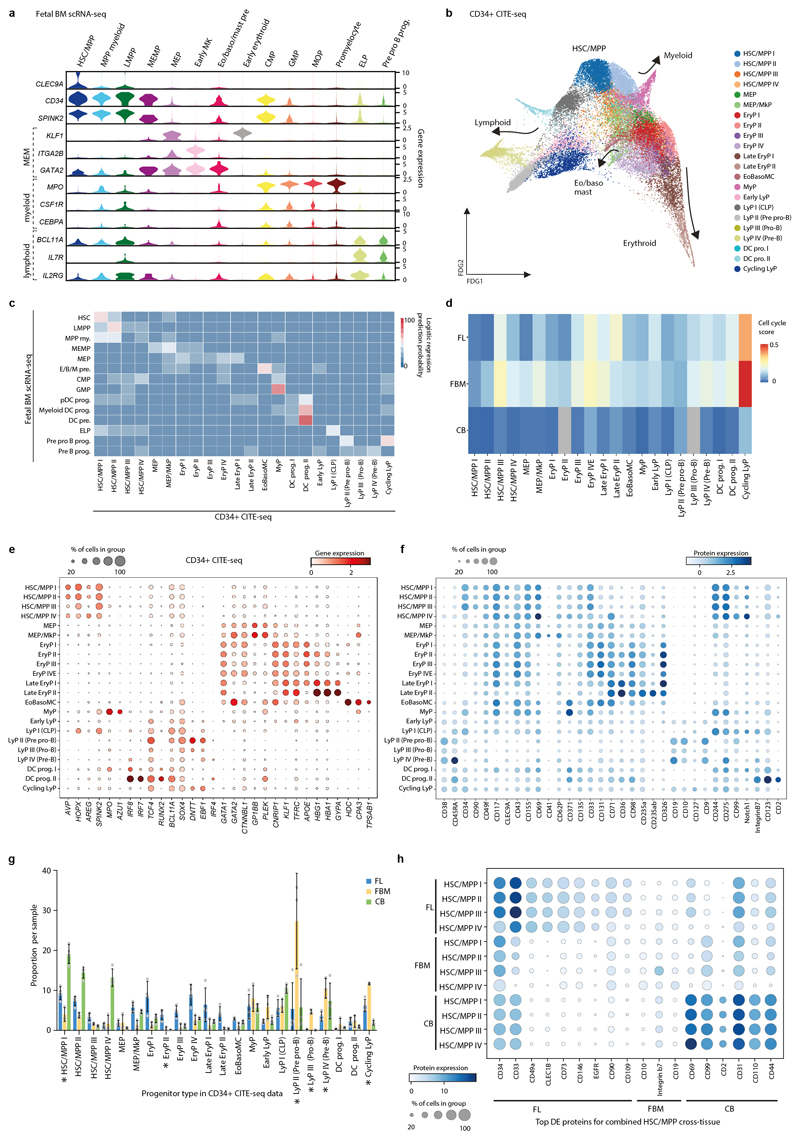
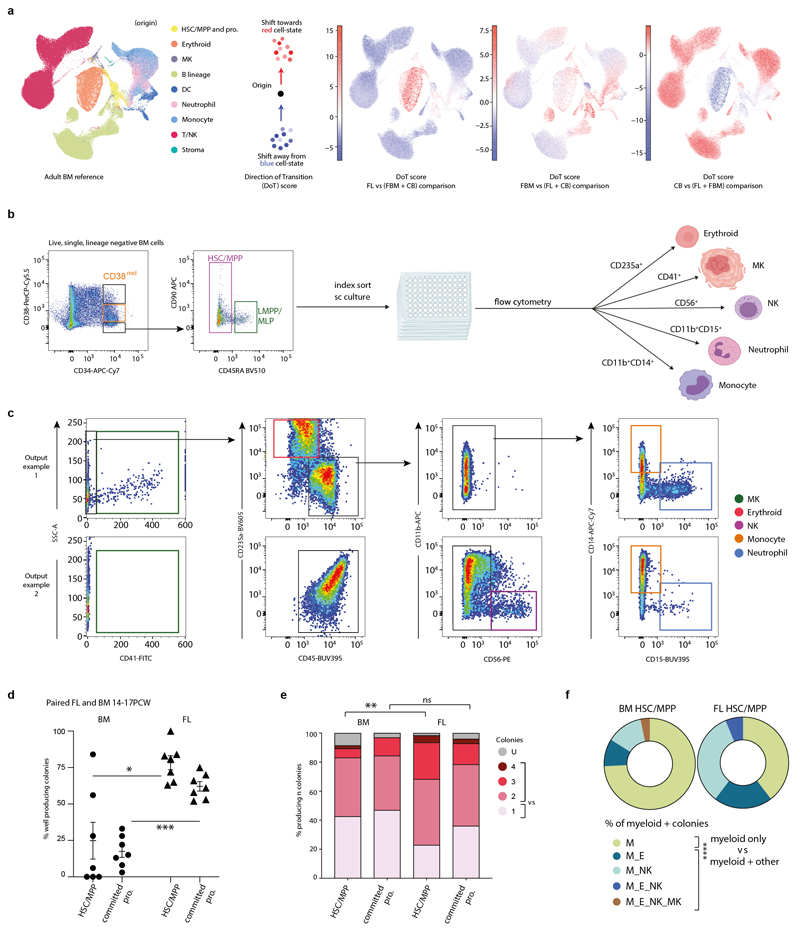
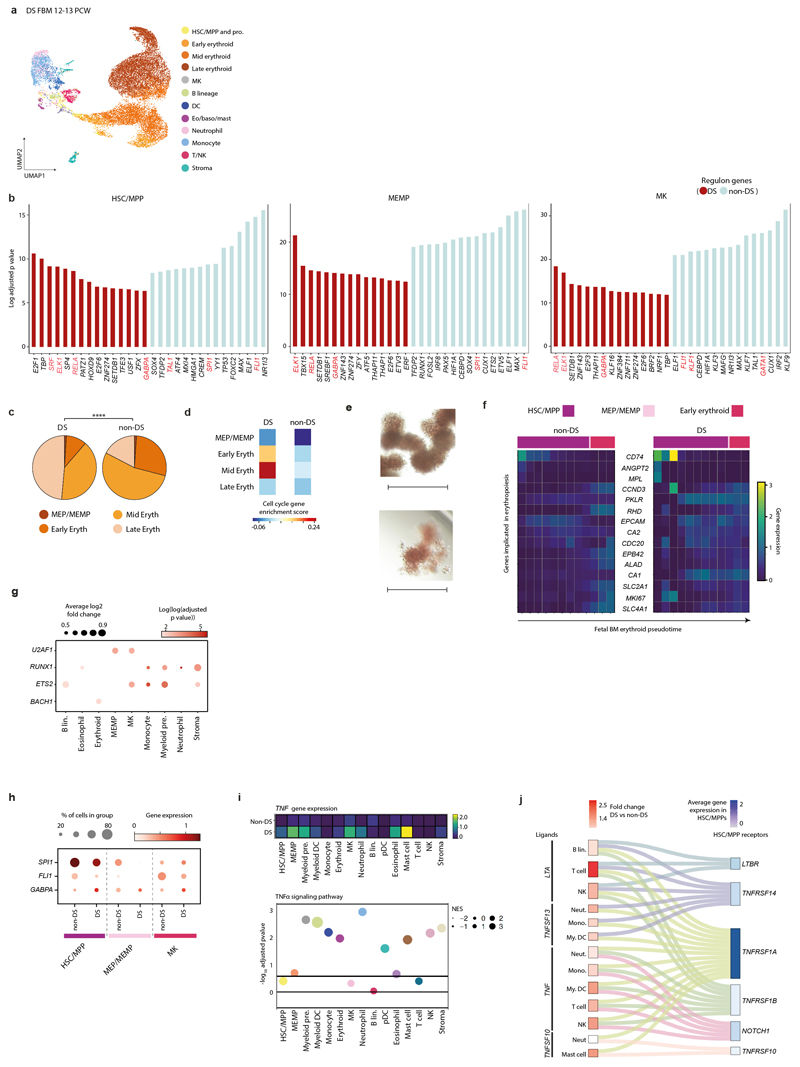
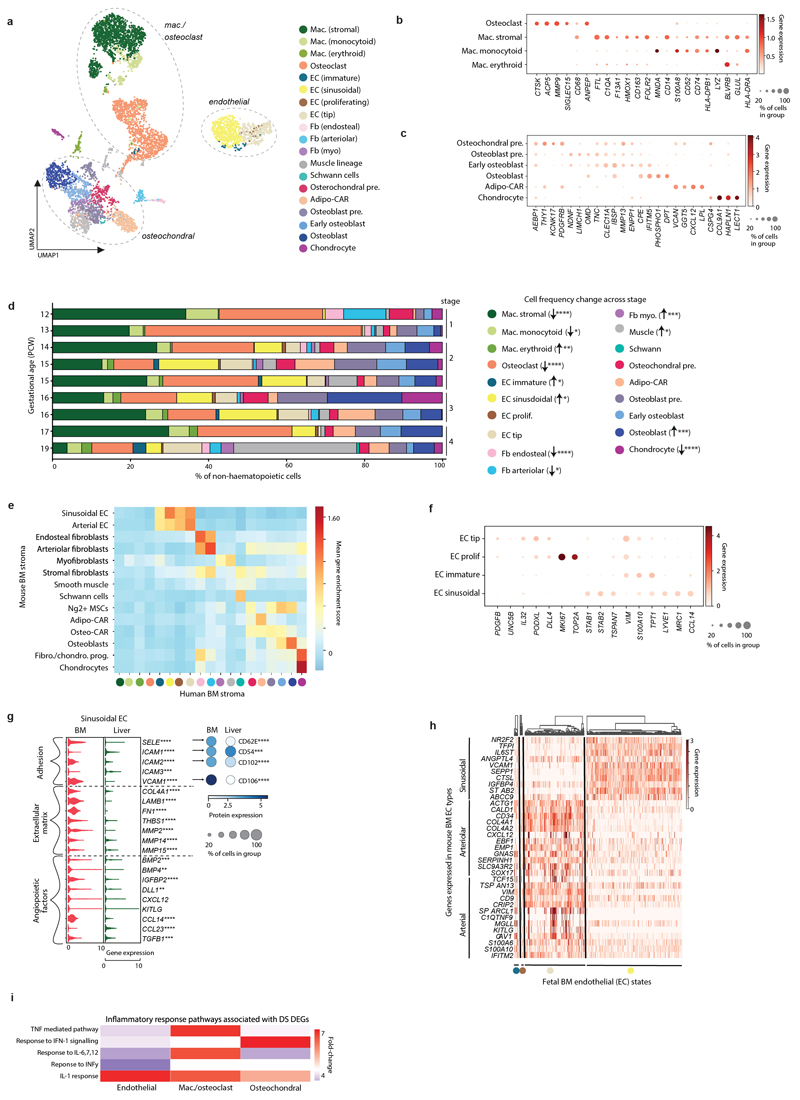
Haematopoiesis in the bone marrow (BM) maintains blood and immune cell production throughout postnatal life. Haematopoiesis first emerges in human BM at 11-12 weeks after conception1,2, yet almost nothing is known about how fetal BM (FBM) evolves to meet the highly specialized needs of the fetus and newborn. Here we detail the development of FBM, including stroma, using multi-omic assessment of mRNA and multiplexed protein epitope expression. We find that the full blood and immune cell repertoire is established in FBM in a short time window of 6-7 weeks early in the second trimester. FBM promotes rapid and extensive diversification of myeloid cells, with granulocytes, eosinophils and dendritic cell subsets emerging for the first time. The substantial expansion of B lymphocytes in FBM contrasts with fetal liver at the same gestational age. Haematopoietic progenitors from fetal liver, FBM and cord blood exhibit transcriptional and functional differences that contribute to tissue-specific identity and cellular diversification. Endothelial cell types form distinct vascular structures that we show are regionally compartmentalized within FBM. Finally, we reveal selective disruption of B lymphocyte, erythroid and myeloid development owing to a cell-intrinsic differentiation bias as well as extrinsic regulation through an altered microenvironment in Down syndrome (trisomy 21).
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
S.O.B is now an employee of Becton, Dickinson and Company (BD); S.O.B's contributions to the work were made prior to the commencement of employment at BD. O.R.R. is an employee of Genentech. O.R.R. is a co-inventor on patent applications filed at the Broad related to single cell genomics. All other authors declare no competing interests.
Figures
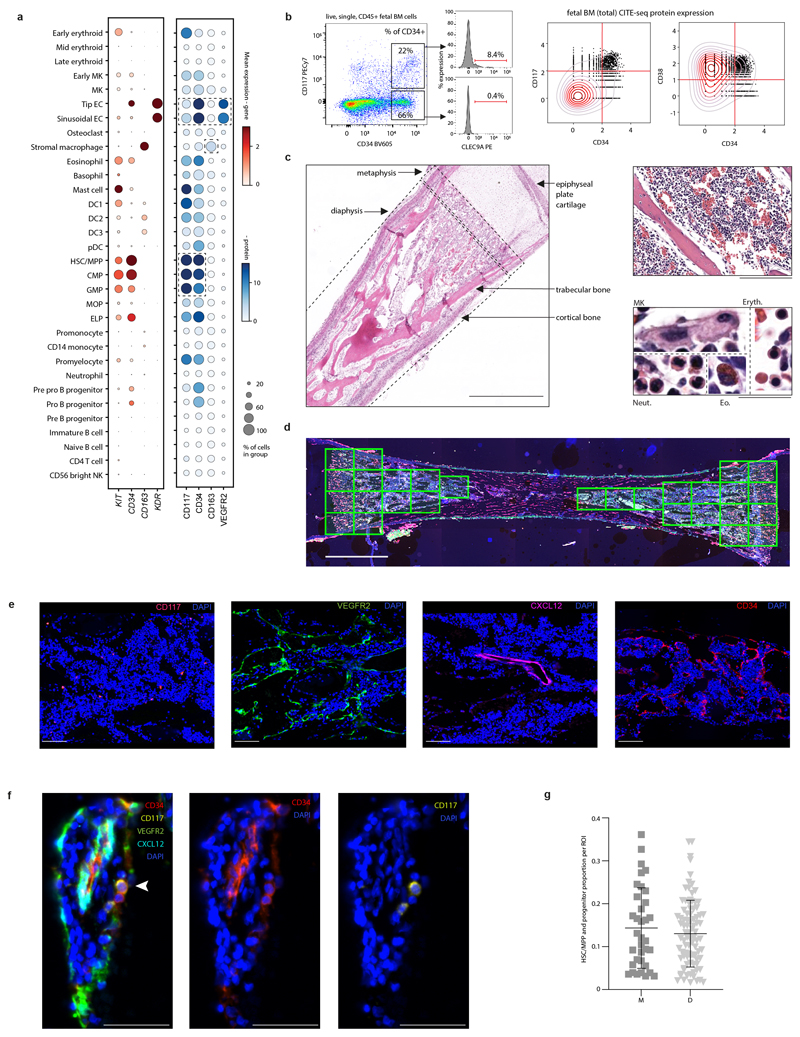
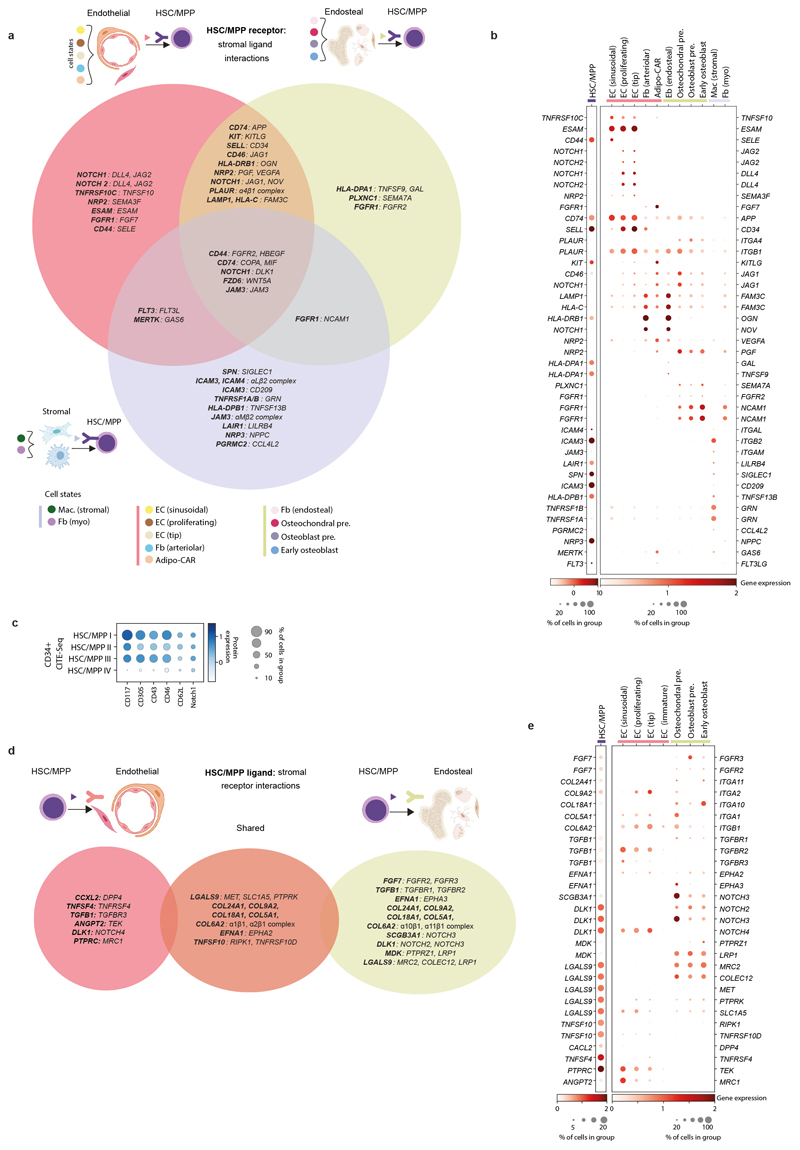

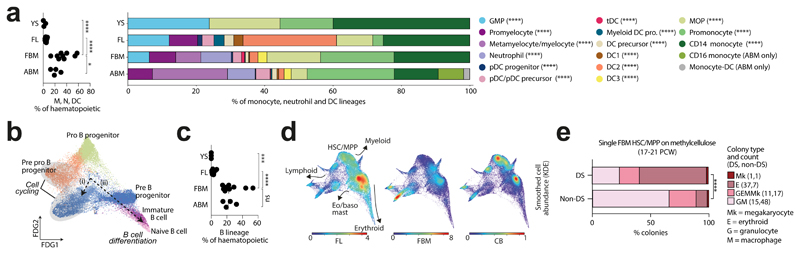
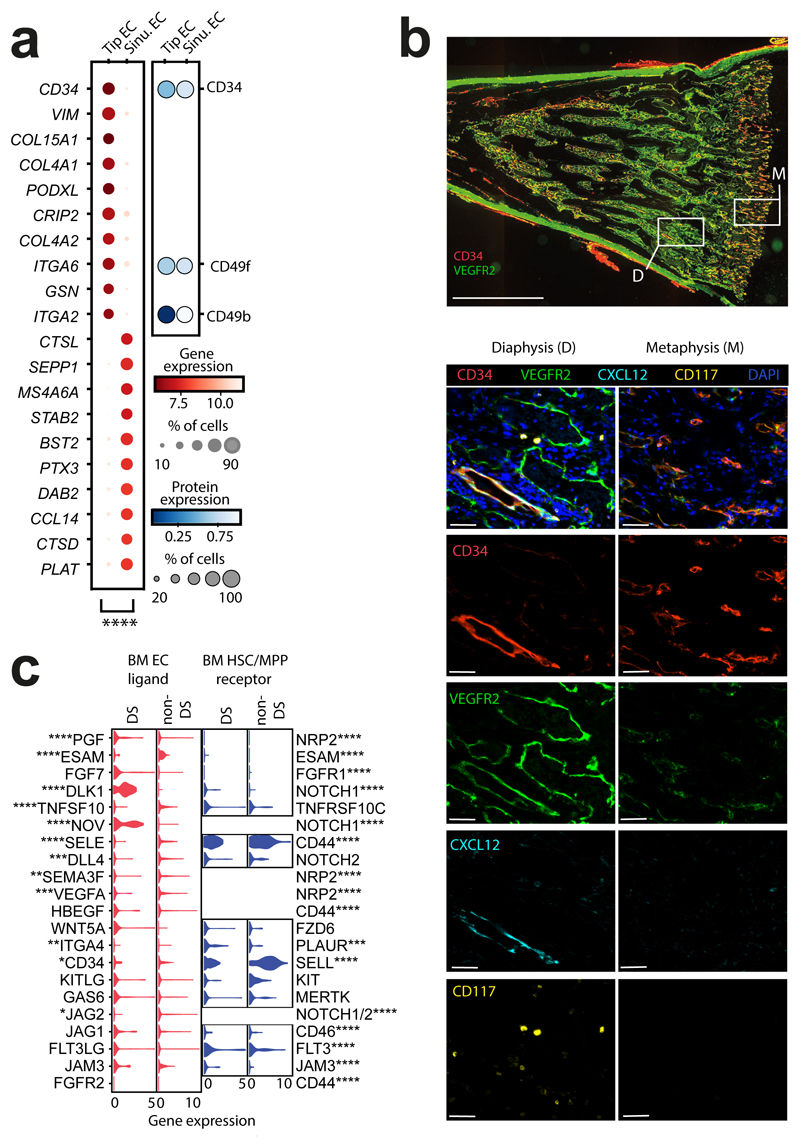
References
-
- O’Byrne S, et al. Discovery of a CD10-negative B-progenitor in human fetal life identifies unique ontogeny-related developmental programs. Blood. 2019;134:1059–1071. - PubMed
-
- Charbord P, Tavian M, Humeau L, Péault B. Early ontogeny of the human marrow from long bones: an immunohistochemical study of hematopoiesis and its microenvironment. Blood. 1996;87:4109–4119. - PubMed
-
- Wiemels JL, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354:1499–1503. - PubMed
Publication types
MeSH terms
Grants and funding
- MC_PC_17230/MRC_/Medical Research Council/United Kingdom
- 107630/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- 216632/WT_/Wellcome Trust/United Kingdom
- 203151/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- 206328/WT_/Wellcome Trust/United Kingdom
- 221052/WT_/Wellcome Trust/United Kingdom
- MR/R006237/1/WTDBT_/DBT-Wellcome Trust India Alliance/India
- 206328/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- MR/S036113/1/MRC_/Medical Research Council/United Kingdom
- 206194/WT_/Wellcome Trust/United Kingdom
- WT107931/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- MR/M008975/1/MRC_/Medical Research Council/United Kingdom
- 107630/WT_/Wellcome Trust/United Kingdom
- MR/W014556/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12009/14/MRC_/Medical Research Council/United Kingdom
- WT206194/WT_/Wellcome Trust/United Kingdom
- 215116/WT_/Wellcome Trust/United Kingdom
- MR/S036334/1/MRC_/Medical Research Council/United Kingdom
- WT211276/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- 216632/Z/19/Z/WT_/Wellcome Trust/United Kingdom
- 107931/WT_/Wellcome Trust/United Kingdom
- 211276/WT_/Wellcome Trust/United Kingdom
- MR/R006237/1/MRC_/Medical Research Council/United Kingdom
- MR/N005872/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
