

MDM-TASK-web: MD-TASK and MODE-TASK web server for analyzing protein dynamics
- PMID: 34589183
- PMCID: PMC8455658
- DOI: 10.1016/j.csbj.2021.08.043
MDM-TASK-web: MD-TASK and MODE-TASK web server for analyzing protein dynamics
Abstract
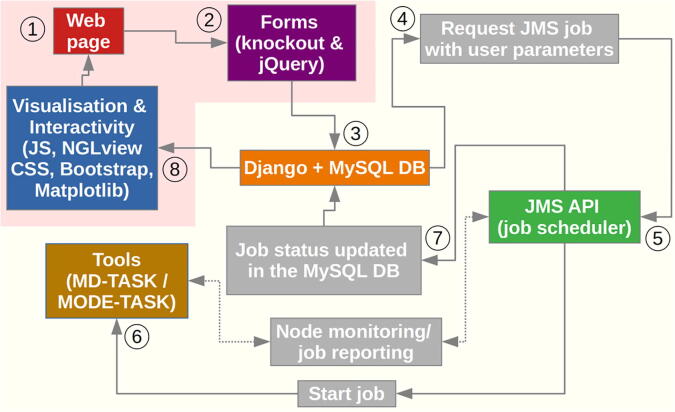
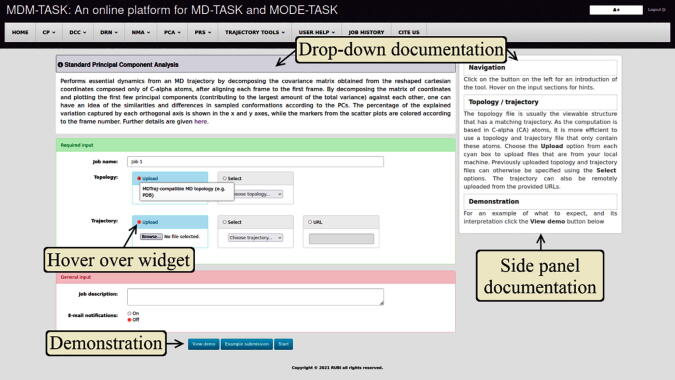
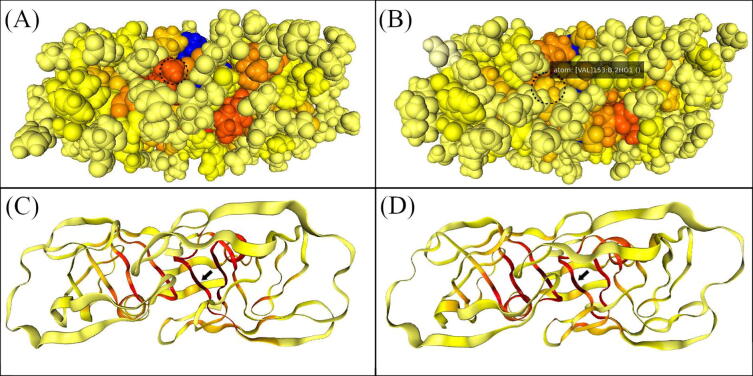
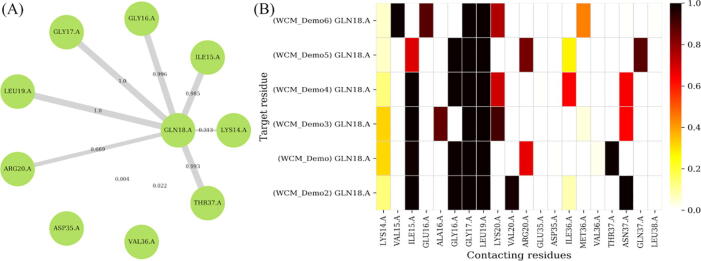
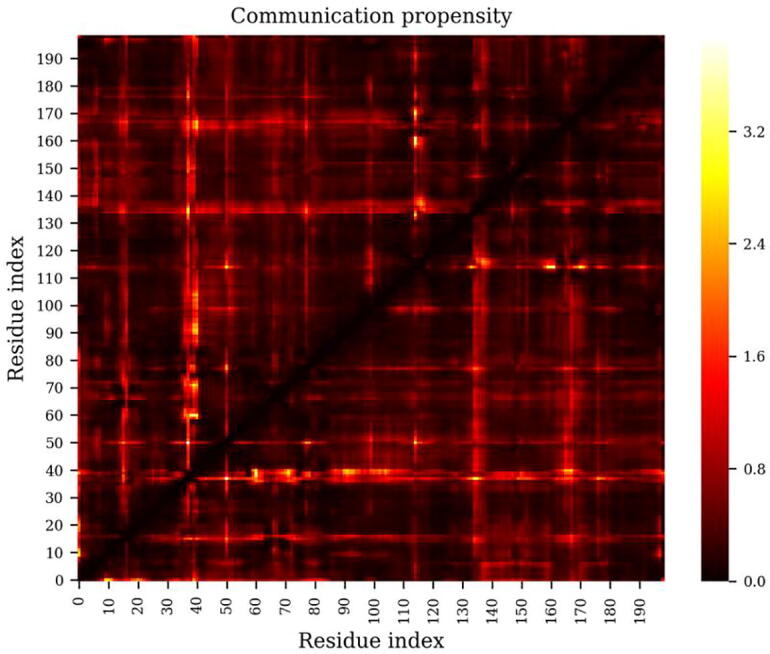
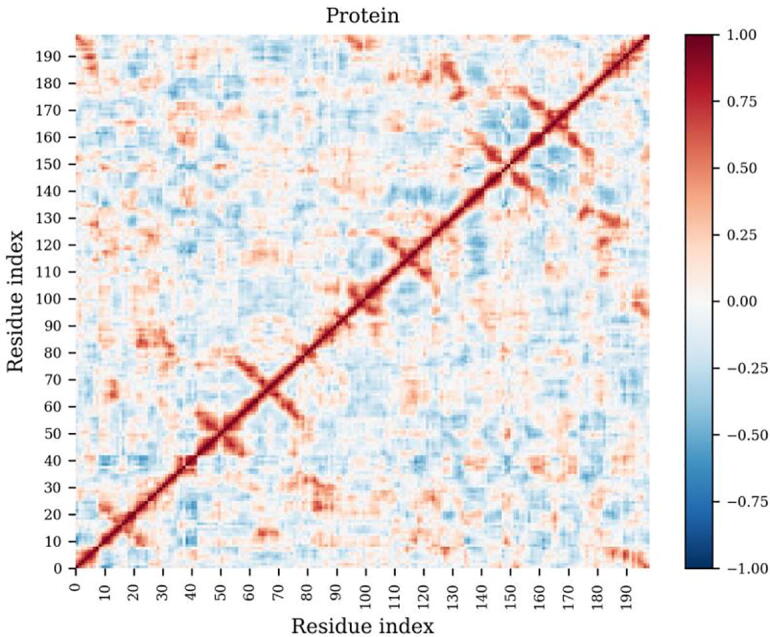
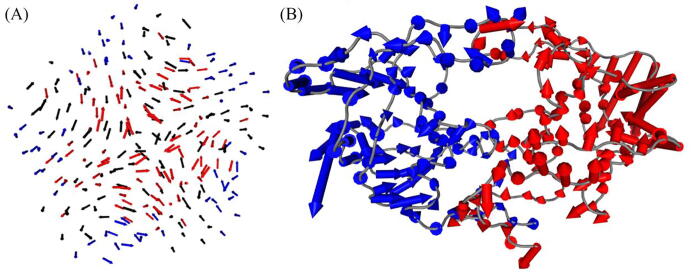
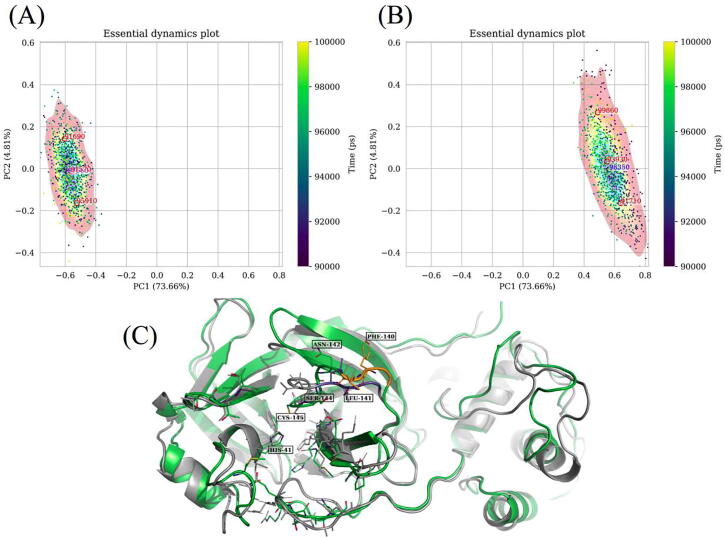
The web server, MDM-TASK-web, combines the MD-TASK and MODE-TASK software suites, which are aimed at the coarse-grained analysis of static and all-atom MD-simulated proteins, using a variety of non-conventional approaches, such as dynamic residue network analysis, perturbation-response scanning, dynamic cross-correlation, essential dynamics and normal mode analysis. Altogether, these tools allow for the exploration of protein dynamics at various levels of detail, spanning single residue perturbations and weighted contact network representations, to global residue centrality measurements and the investigation of global protein motion. Typically, following molecular dynamic simulations designed to investigate intrinsic and extrinsic protein perturbations (for instance induced by allosteric and orthosteric ligands, protein binding, temperature, pH and mutations), this selection of tools can be used to further describe protein dynamics. This may lead to the discovery of key residues involved in biological processes, such as drug resistance. The server simplifies the set-up required for running these tools and visualizing their results. Several scripts from the tool suites were updated and new ones were also added and integrated with 2D/3D visualization via the web interface. An embedded work-flow, integrated documentation and visualization tools shorten the number of steps to follow, starting from calculations to result visualization. The Django-powered web server (available at https://mdmtaskweb.rubi.ru.ac.za/) is compatible with all major web browsers. All scripts implemented in the web platform are freely available at https://github.com/RUBi-ZA/MD-TASK/tree/mdm-task-web and https://github.com/RUBi-ZA/MODE-TASK/tree/mdm-task-web.
Keywords: MD-TASK; MODE-TASK; Molecular dynamics analysis; Normal mode analysis; Residue network analysis.
© 2021 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Golbeck J. Newnes; 2013. Analyzing the social web.
-
- Contreras-Riquelme S, Garate J-A, Perez-Acle T, Martin AJM, RIP-MD: a tool to study residue interaction networks in protein molecular dynamics, PeerJ. 6 (2018) e5998. https://doi.org/10.7717/peerj.5998. - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources