

Mechanisms of UV-induced mutations and skin cancer
- PMID: 34589668
- PMCID: PMC8477449
- DOI: 10.1007/s42764-020-00009-8
Mechanisms of UV-induced mutations and skin cancer
Abstract
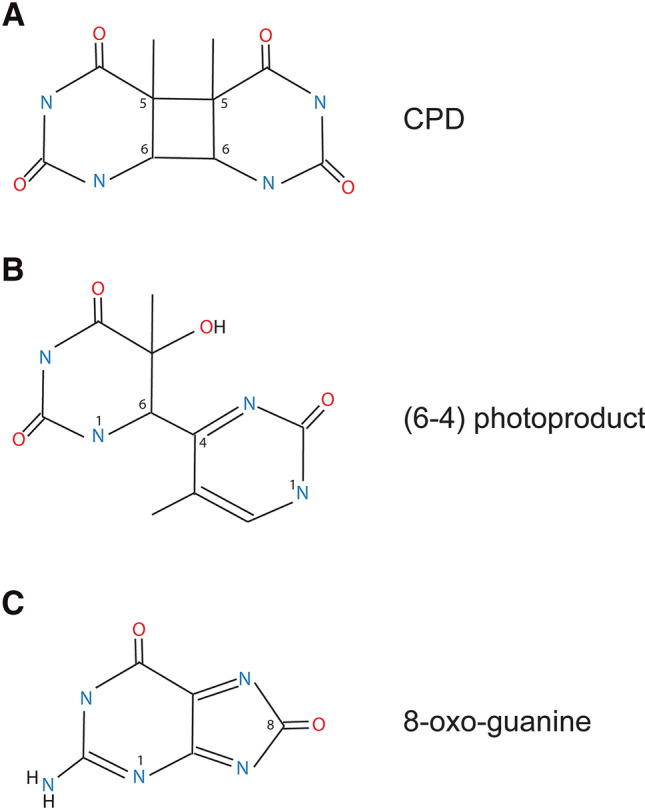
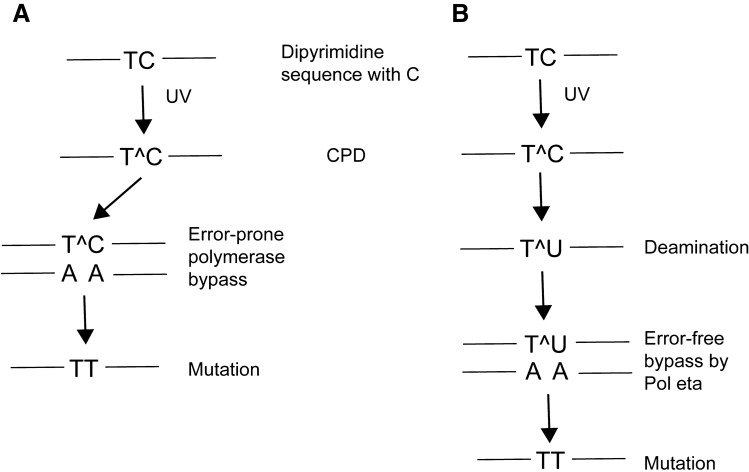
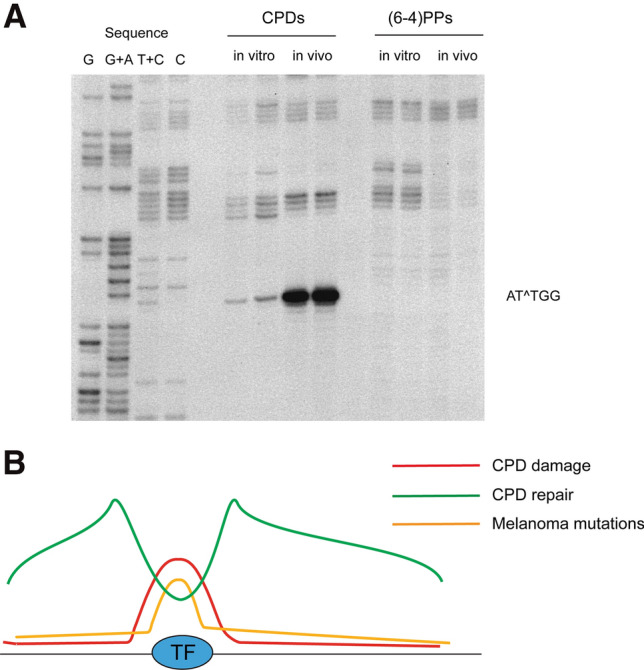
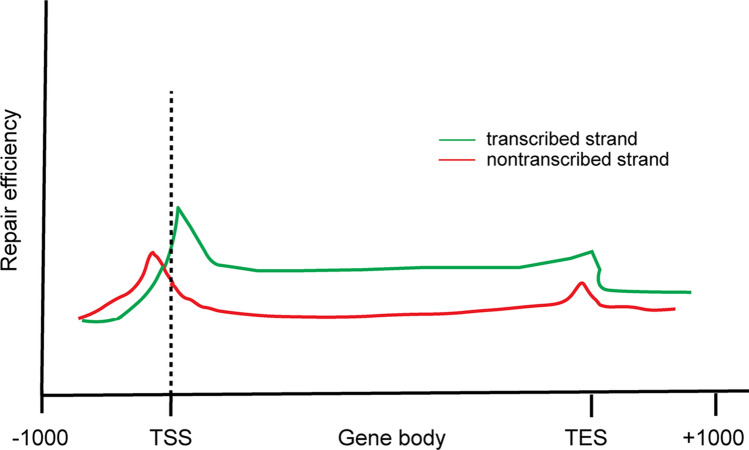
Ultraviolet (UV) irradiation causes various types of DNA damage, which leads to specific mutations and the emergence of skin cancer in humans, often decades after initial exposure. Different UV wavelengths cause the formation of prominent UV-induced DNA lesions. Most of these lesions are removed by the nucleotide excision repair pathway, which is defective in rare genetic skin disorders referred to as xeroderma pigmentosum. A major role in inducing sunlight-dependent skin cancer mutations is assigned to the cyclobutane pyrimidine dimers (CPDs). In this review, we discuss the mechanisms of UV damage induction, the genomic distribution of this damage, relevant DNA repair mechanisms, the proposed mechanisms of how UV-induced CPDs bring about DNA replication-dependent mutagenicity in mammalian cells, and the strong signature of UV damage and mutagenesis found in skin cancer genomes.
Keywords: (6–4) photoproduct; DNA repair; UV; Ultraviolet; cyclobutane pyrimidine dimer; melanoma; mutations; skin cancer.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources