

Principles of mRNA targeting via the Arabidopsis m6A-binding protein ECT2
- PMID: 34591015
- PMCID: PMC8796052
- DOI: 10.7554/eLife.72375
Principles of mRNA targeting via the Arabidopsis m6A-binding protein ECT2
Abstract
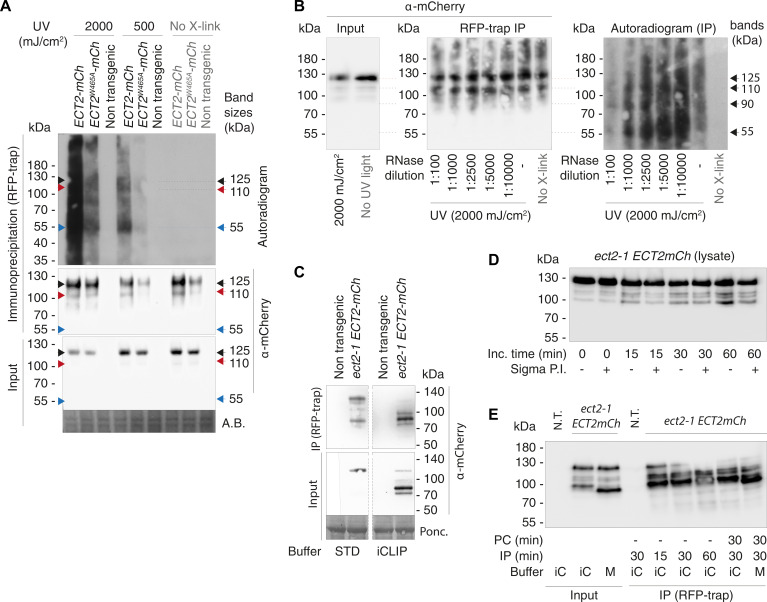
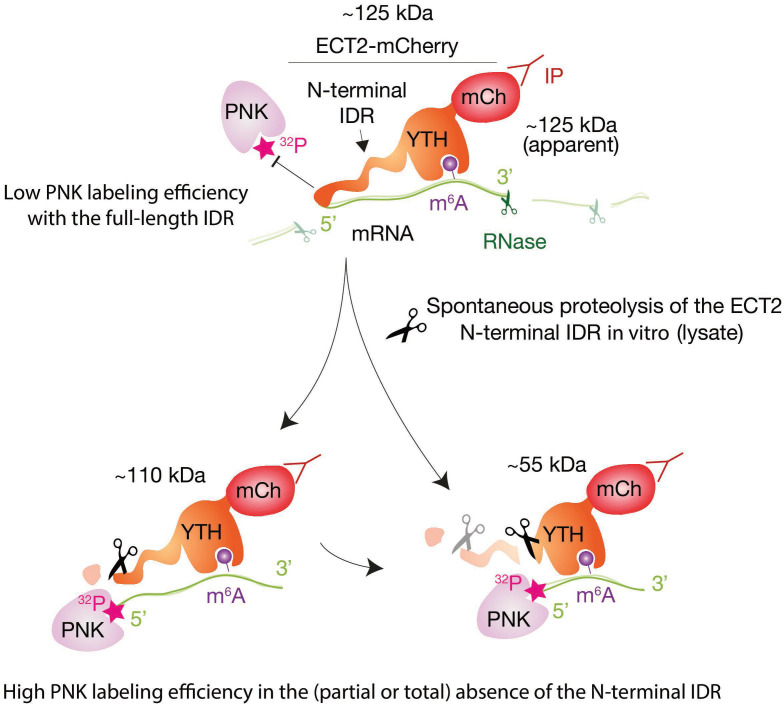
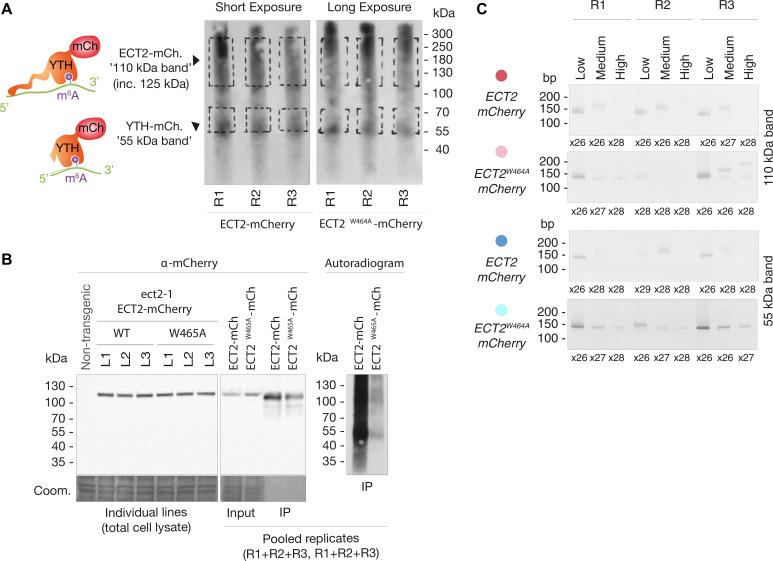
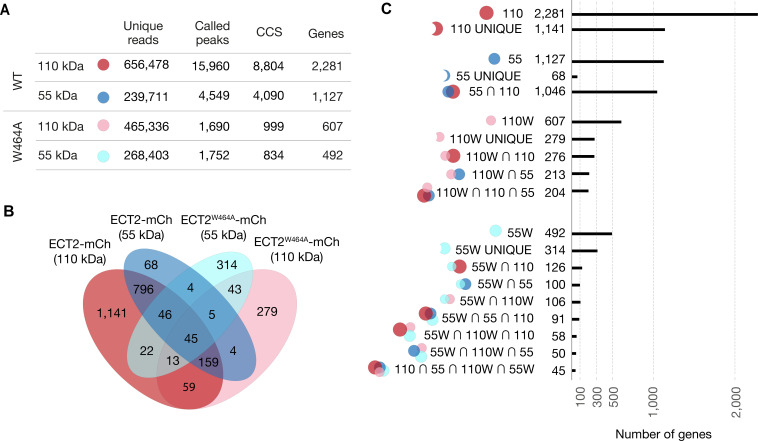
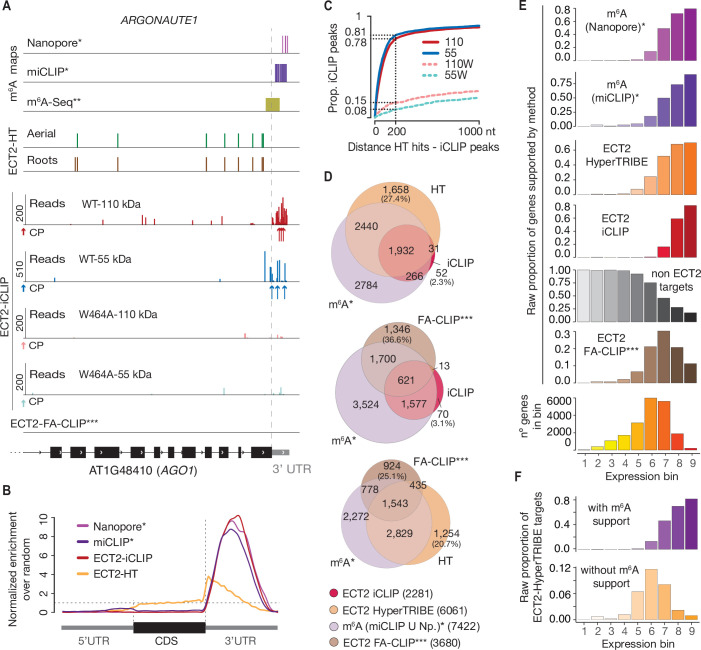
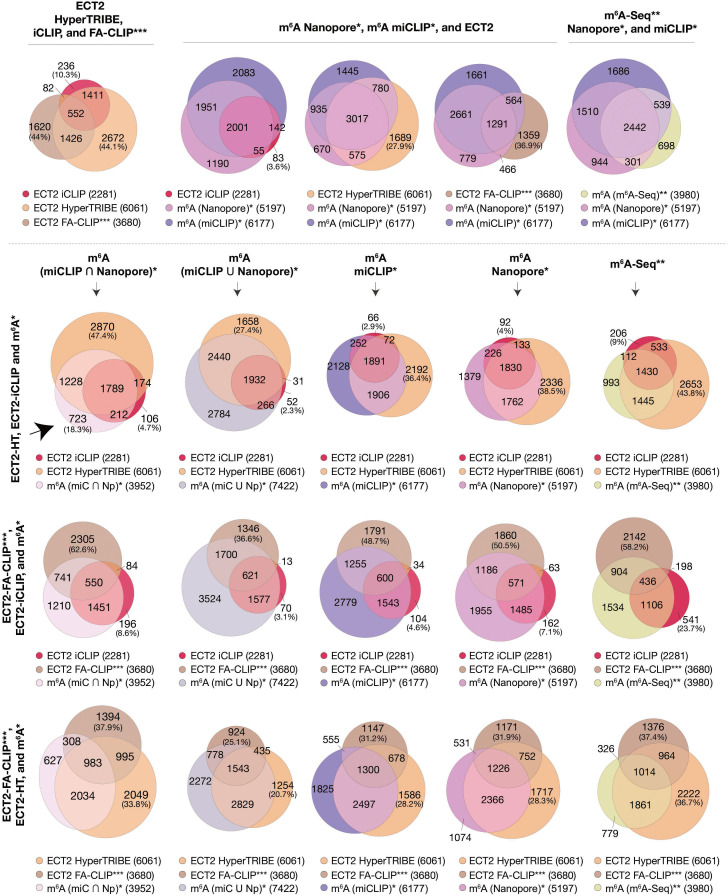
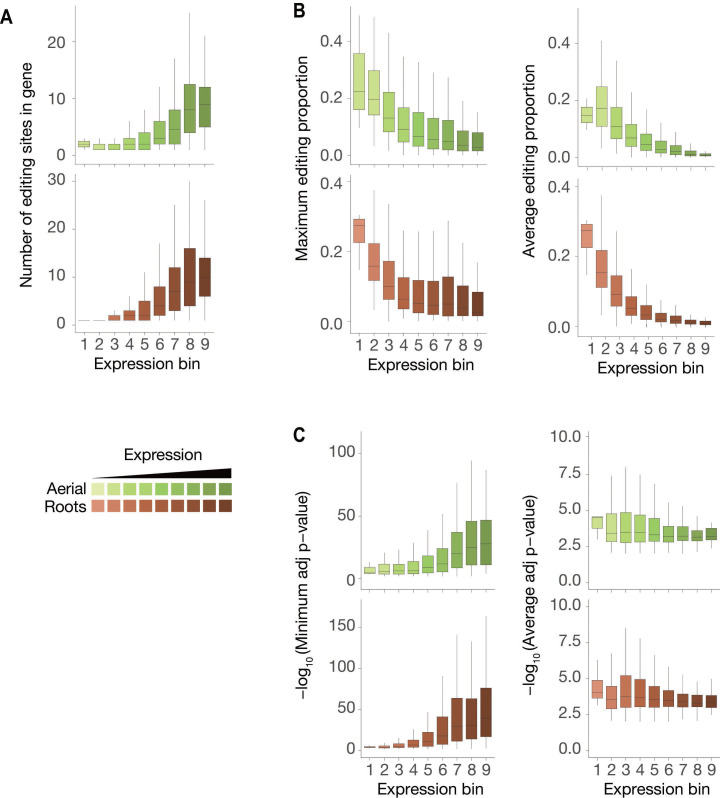
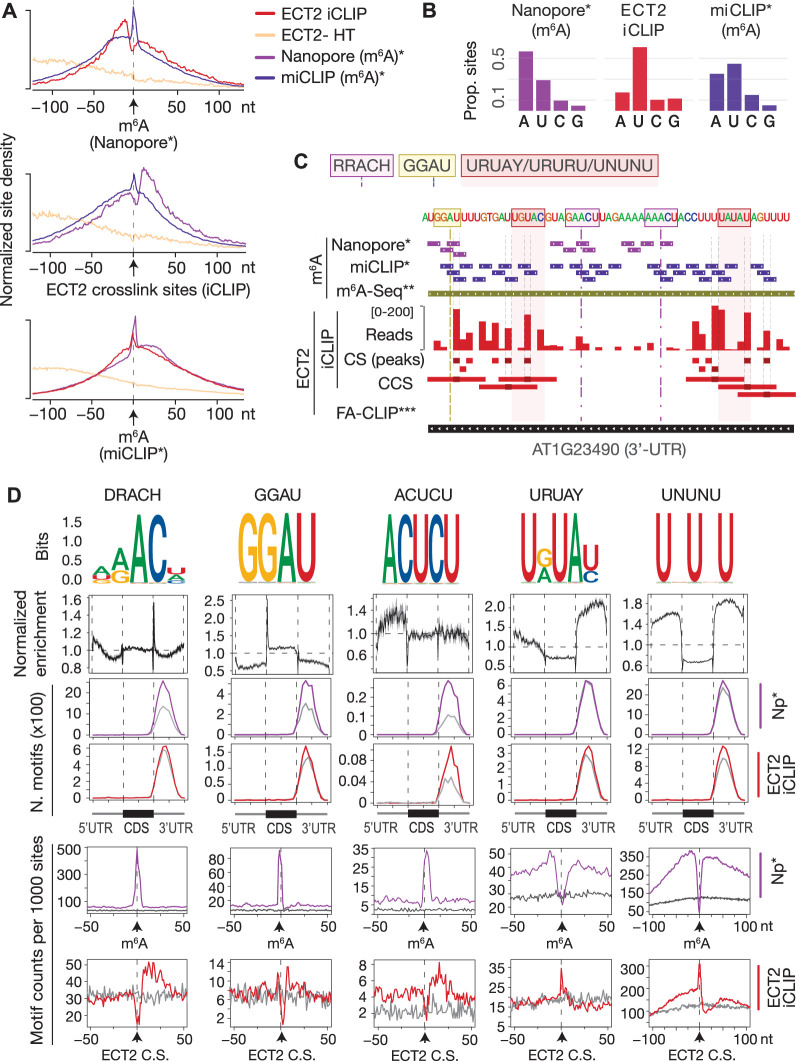
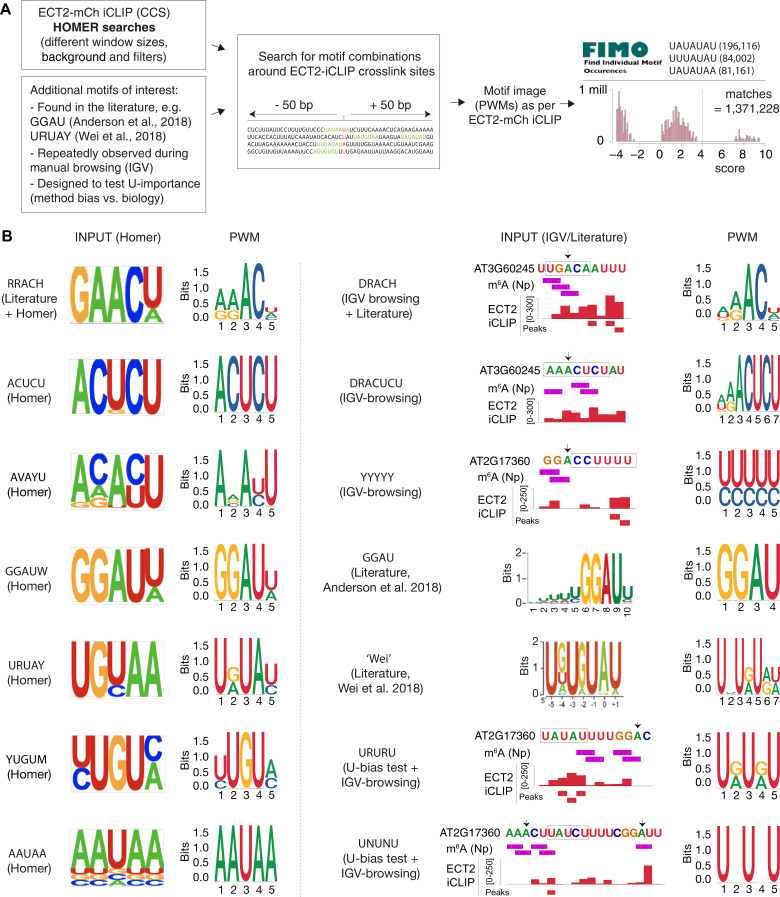
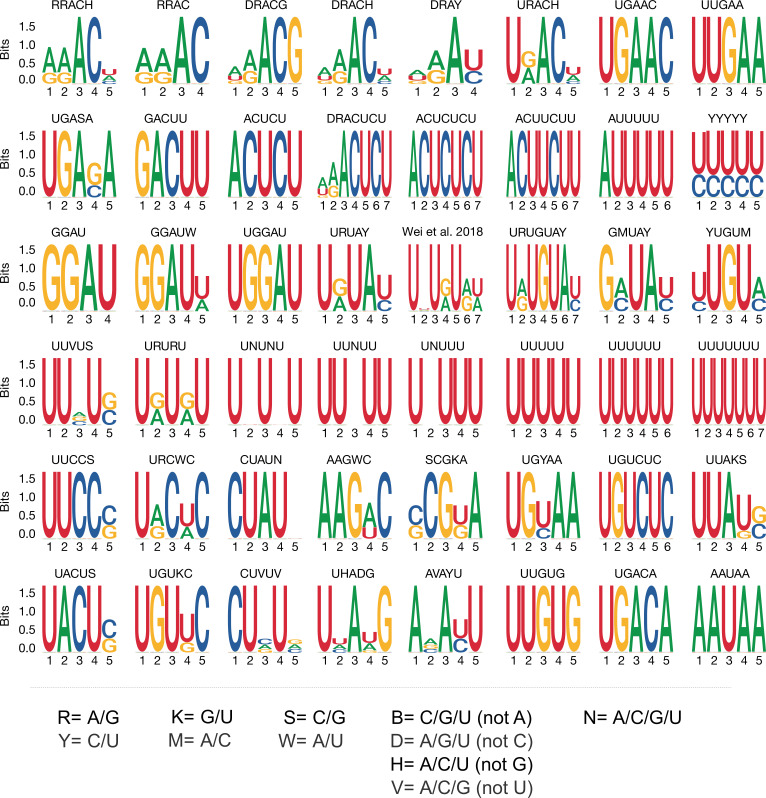
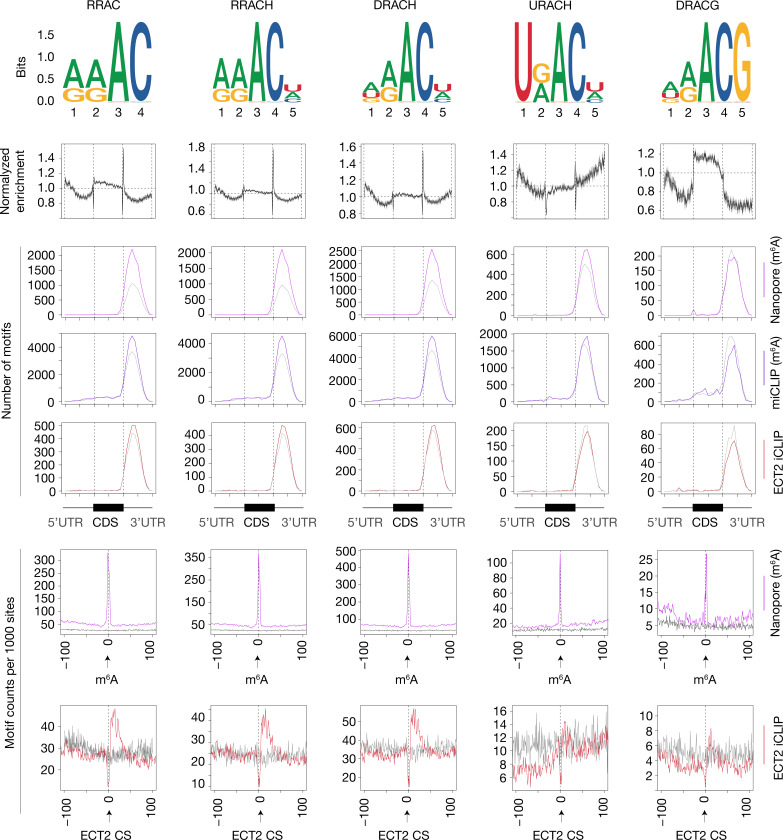
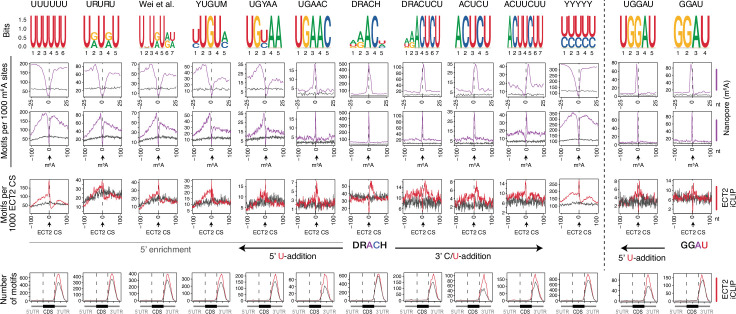
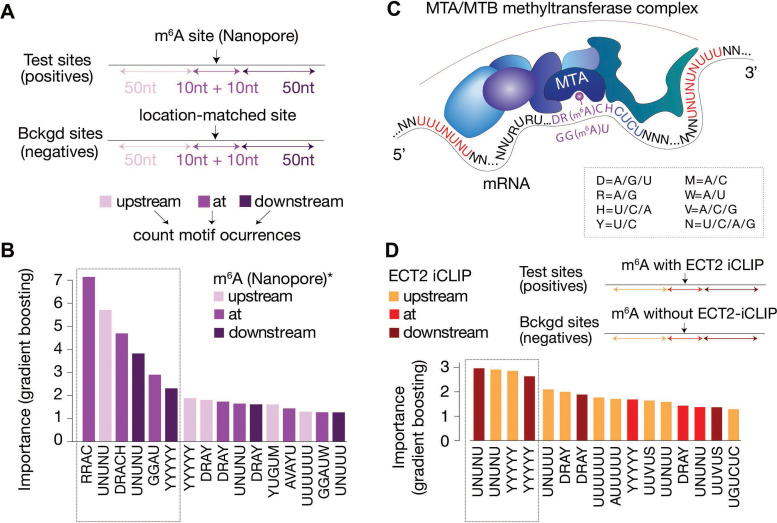
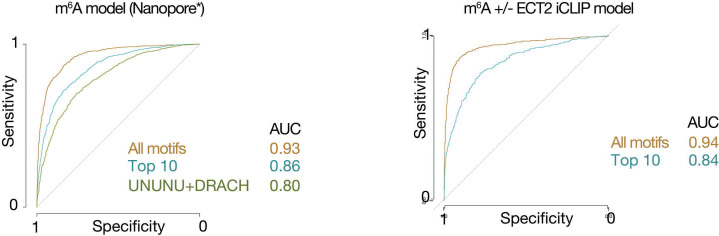
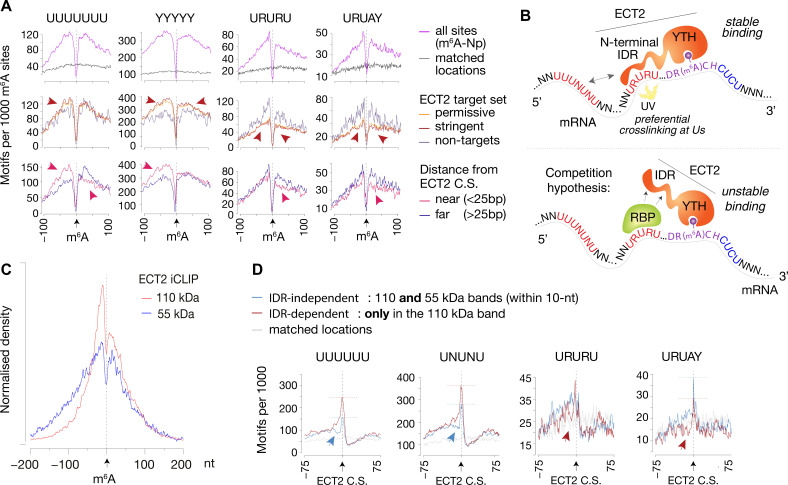
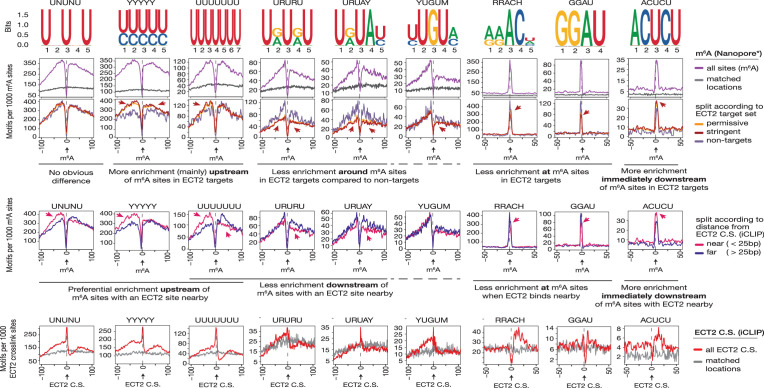
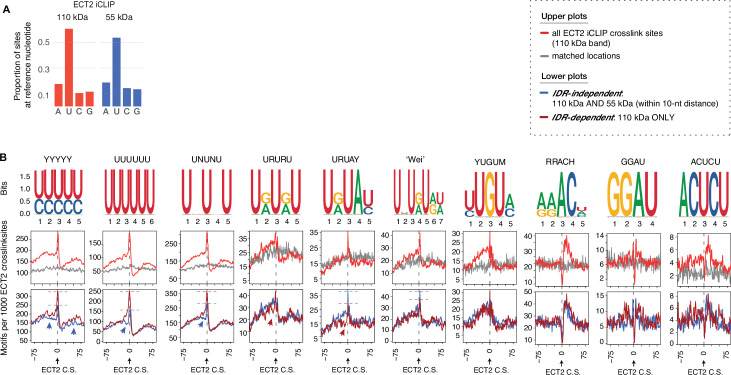
Specific recognition of N6-methyladenosine (m6A) in mRNA by RNA-binding proteins containing a YT521-B homology (YTH) domain is important in eukaryotic gene regulation. The Arabidopsis YTH domain protein ECT2 is thought to bind to mRNA at URU(m6A)Y sites, yet RR(m6A)CH is the canonical m6A consensus site in all eukaryotes and ECT2 functions require m6A-binding activity. Here, we apply iCLIP (individual nucleotide resolution crosslinking and immunoprecipitation) and HyperTRIBE (targets of RNA-binding proteins identified by editing) to define high-quality target sets of ECT2 and analyze the patterns of enriched sequence motifs around ECT2 crosslink sites. Our analyses show that ECT2 does in fact bind to RR(m6A)CH. Pyrimidine-rich motifs are enriched around, but not at m6A sites, reflecting a preference for N6-adenosine methylation of RRACH/GGAU islands in pyrimidine-rich regions. Such motifs, particularly oligo-U and UNUNU upstream of m6A sites, are also implicated in ECT2 binding via its intrinsically disordered region (IDR). Finally, URUAY-type motifs are enriched at ECT2 crosslink sites, but their distinct properties suggest function as sites of competition between binding of ECT2 and as yet unidentified RNA-binding proteins. Our study provides coherence between genetic and molecular studies of m6A-YTH function in plants and reveals new insight into the mode of RNA recognition by YTH domain-containing proteins.
Keywords: A. thaliana; ECT2; GGAU; RRACH; URUAY; genetics; genomics; hyperTRIBE; iCLIP; m6A; motif; plant biology; target.
Plain language summary
Genes are strings of genetic code that contain instructions for producing a cell’s proteins. Active genes are copied from DNA into molecules called mRNAs, and mRNA molecules are subsequently translated to create new proteins. However, the number of proteins produced by a cell is not only limited by the number of mRNA molecules produced by copying DNA. Cells use a variety of methods to control the stability of mRNA molecules and their translation efficiency to regulate protein production. One of these methods involves adding a chemical tag, a methyl group, onto mRNA while it is being created. These methyl tags can then be used as docking stations by RNA-binding proteins that help regulate protein translation. Most eukaryotic species – which include animals, plants and fungi – use the same system to add methyl tags to mRNA molecules. One methyl tag in particular, known as m6A, is a well-characterised docking site for a particular type of RNA-binding protein that goes by the name of ECT2 in plants. However, in the flowering plant Arabidopsis thaliana, ECT2 was thought to bind to an mRNA sequence different from the one normally carrying the chemical tag, creating obvious confusion about how the system works in plants. Arribas-Hernández, Rennie et al. investigated this question using advanced large-scale biochemical techniques, and discovered that conventional m6A methyl tags are indeed used by ECT2 in Arabidopsis thaliana. The confusion likely arose because the sequence ECT2 was thought bind is often located in close proximity to the m6A tags, possibly acting as docking stations for proteins that can influence the ability of ECT2 to bind mRNA. Arribas-Hernández, Rennie et al. also uncovered additional mRNA sequences that directly interact with parts of ECT2 previously unknown to participate in mRNA binding. These findings provide new insights into how chemical labels in mRNA control gene activity. They have broad implications that extend beyond plants into other eukaryotic species, including humans. Since this chemical labelling system has a major role in controlling plant growth, these findings could be leveraged in biotechnology applications to improve crop yields and enhance plant-based food production.
© 2021, Arribas-Hernández et al.
Conflict of interest statement
LA, SR, TK, CP, ML, DS, RA, PB No competing interests declared
Figures
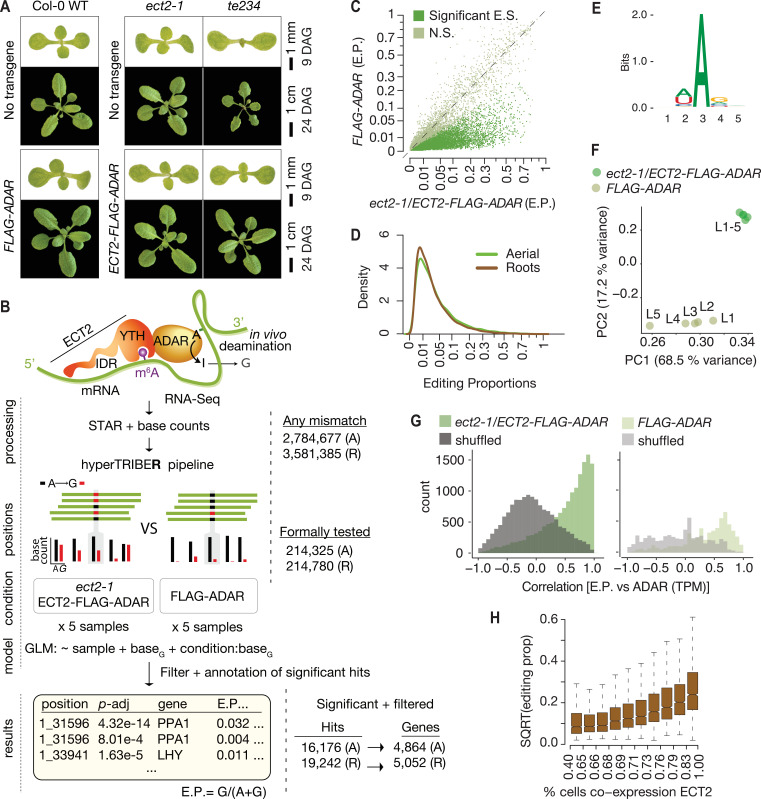
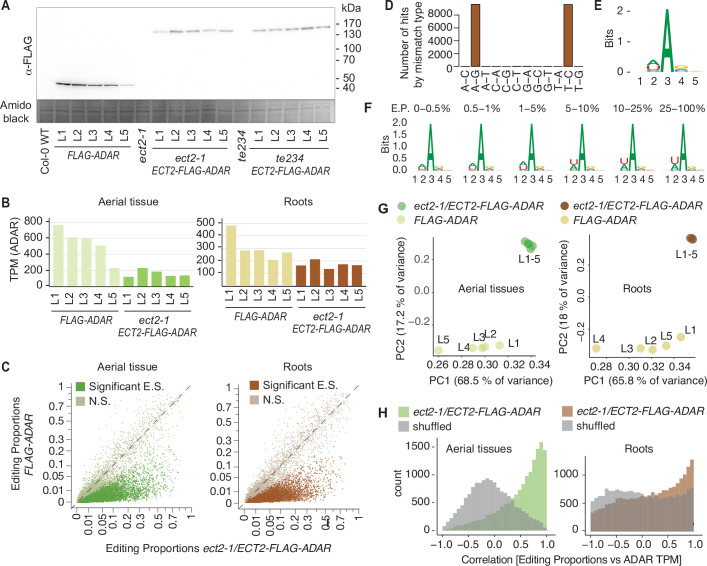
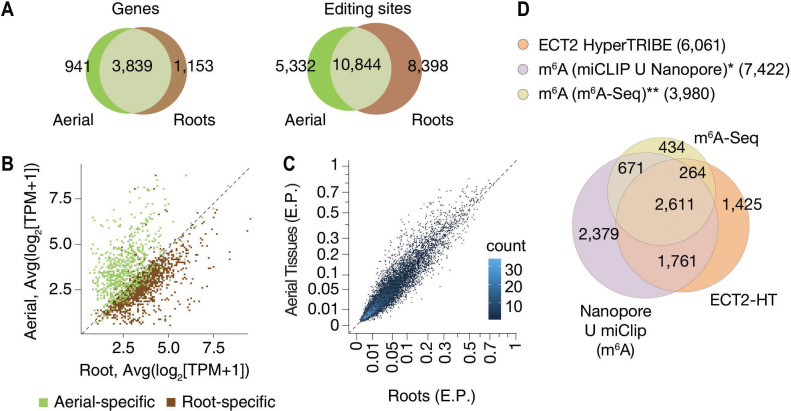
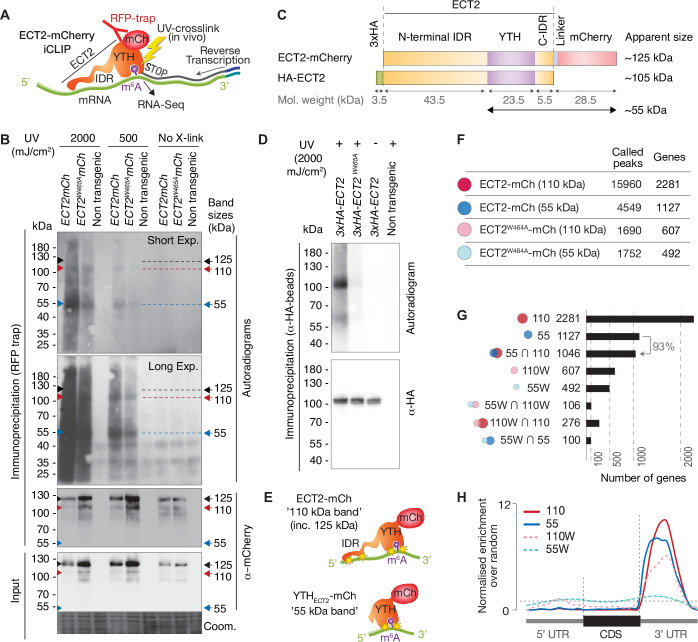

References
-
- Arribas-Hernández L, Marchais A, Poulsen C, Haase B, Hauptmann J, Benes V, Meister G, Brodersen P. The slicer activity of ARGONAUTE1 is required specifically for the phasing, not production, of trans-acting short interfering RNAs in Arabidopsis. The Plant Cell. 2016;28:1563–1580. doi: 10.1105/tpc.16.00121. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
