

Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states
- PMID: 34594037
- PMCID: PMC8675181
- DOI: 10.1038/s41588-021-00927-7
Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states
Abstract
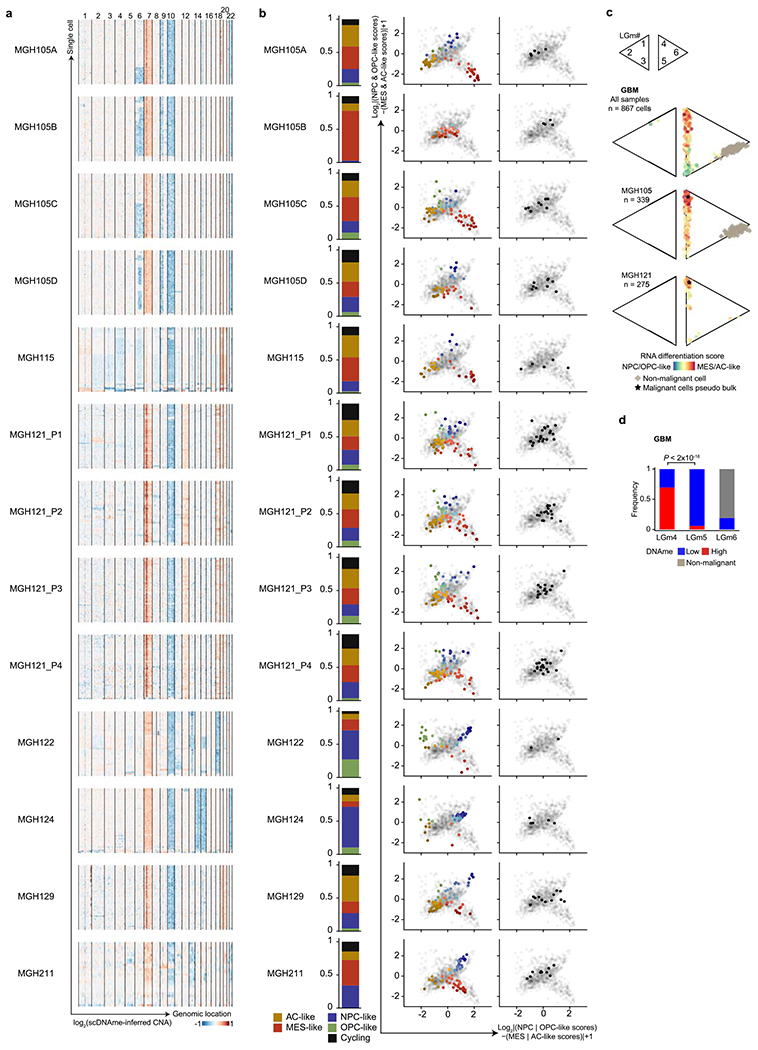
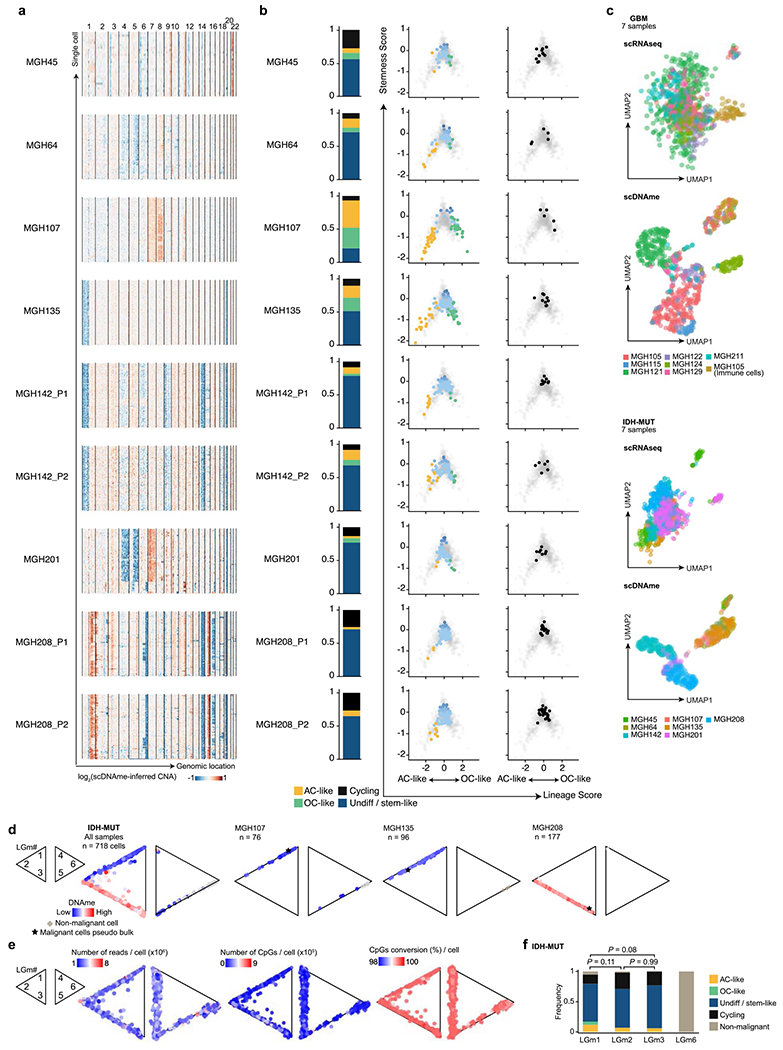
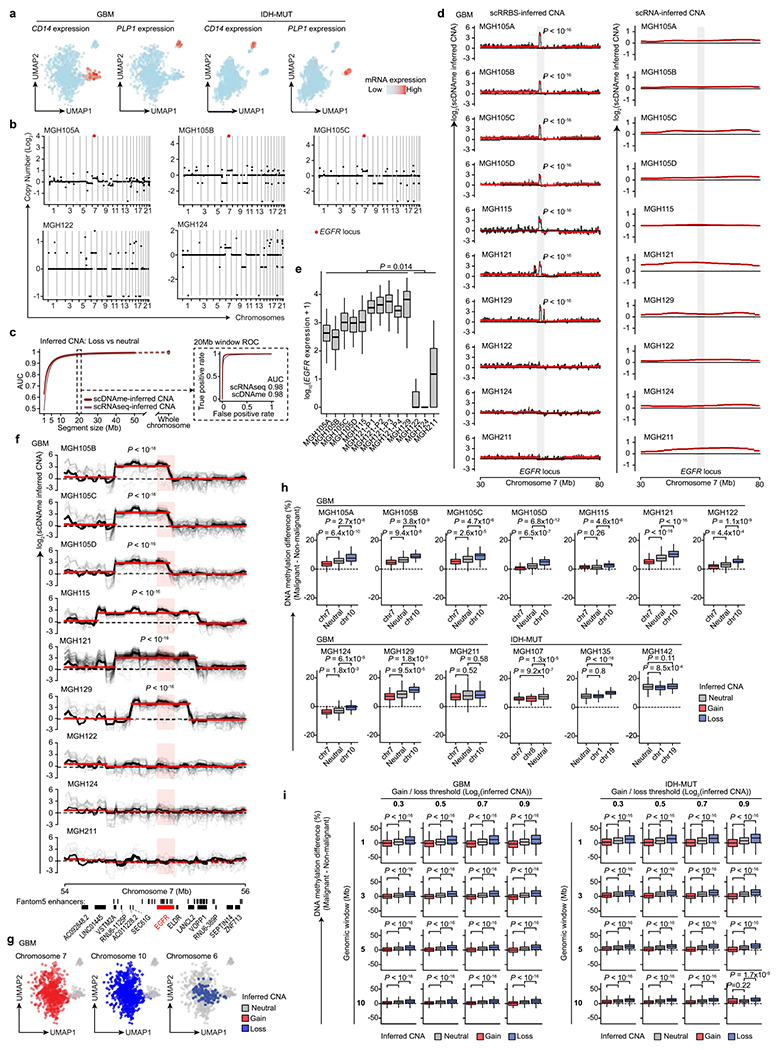
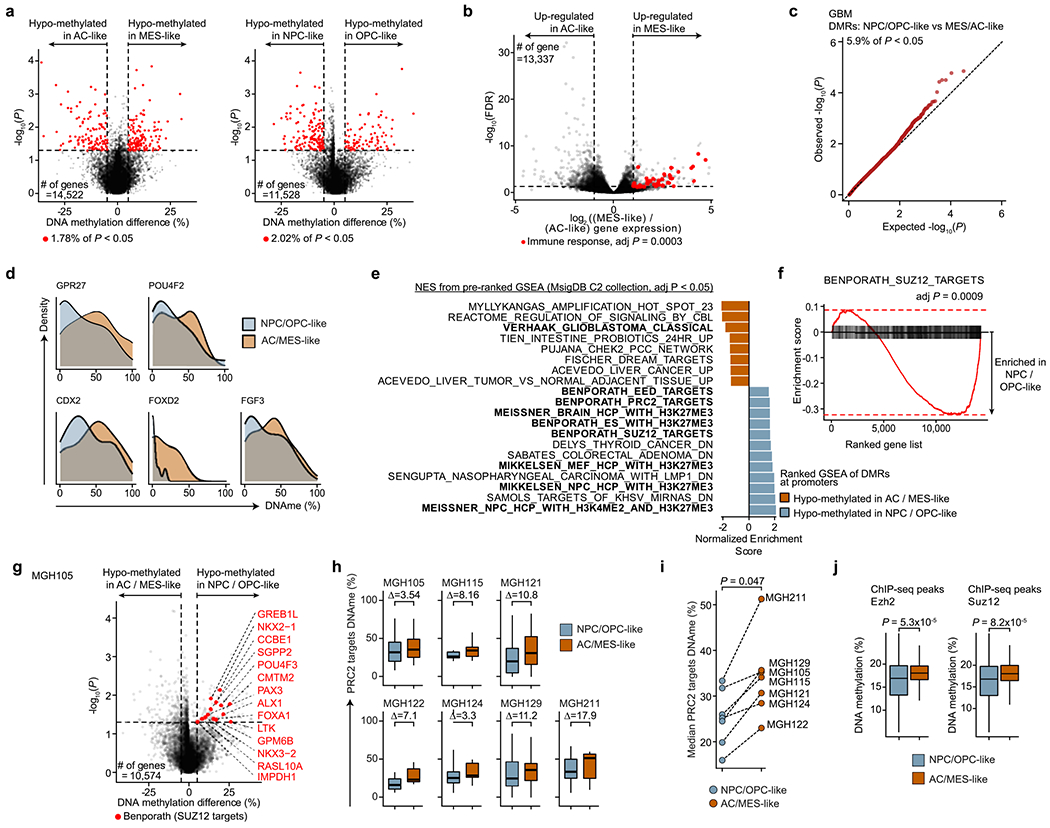
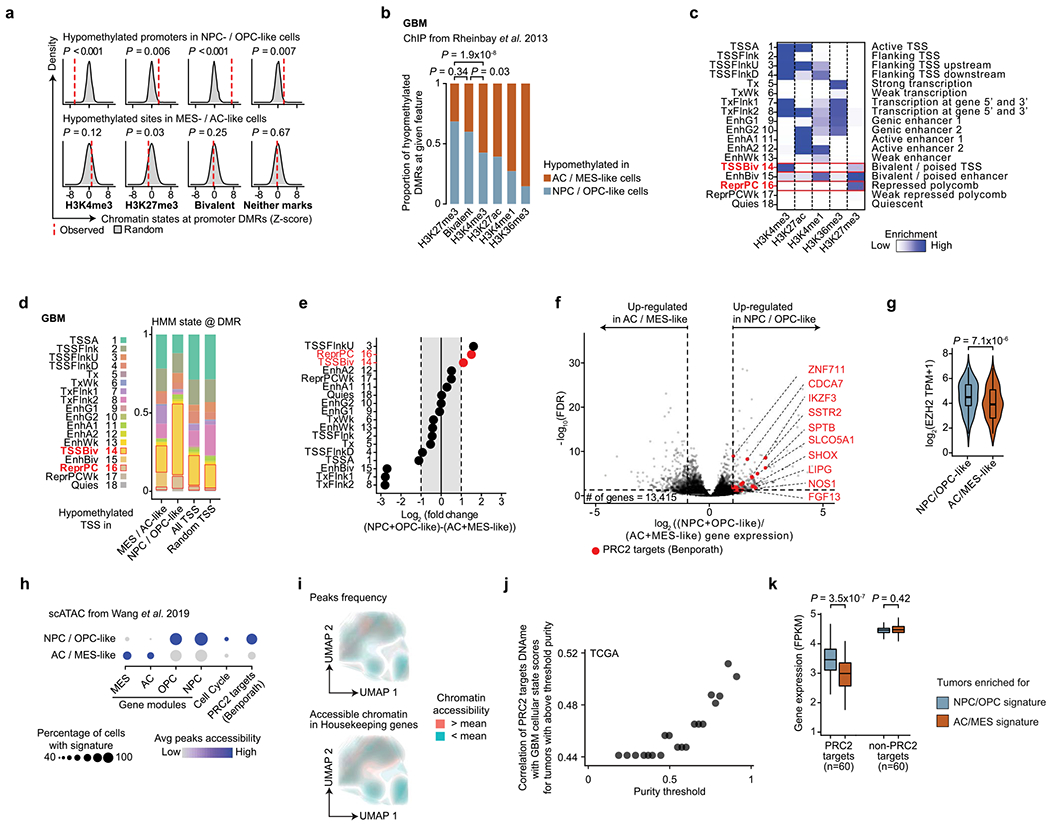
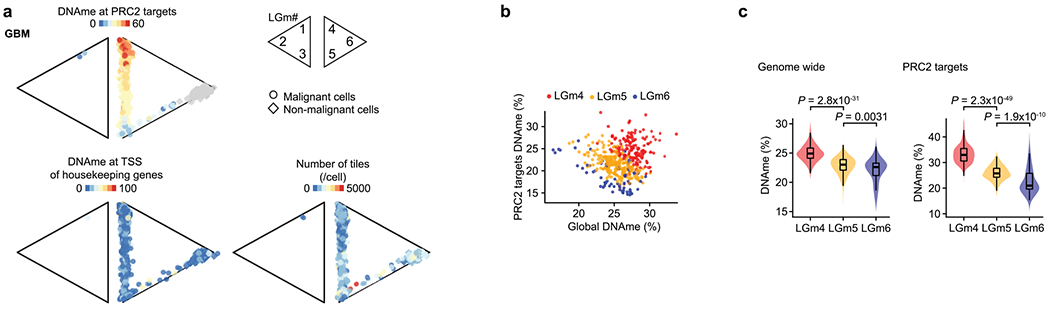
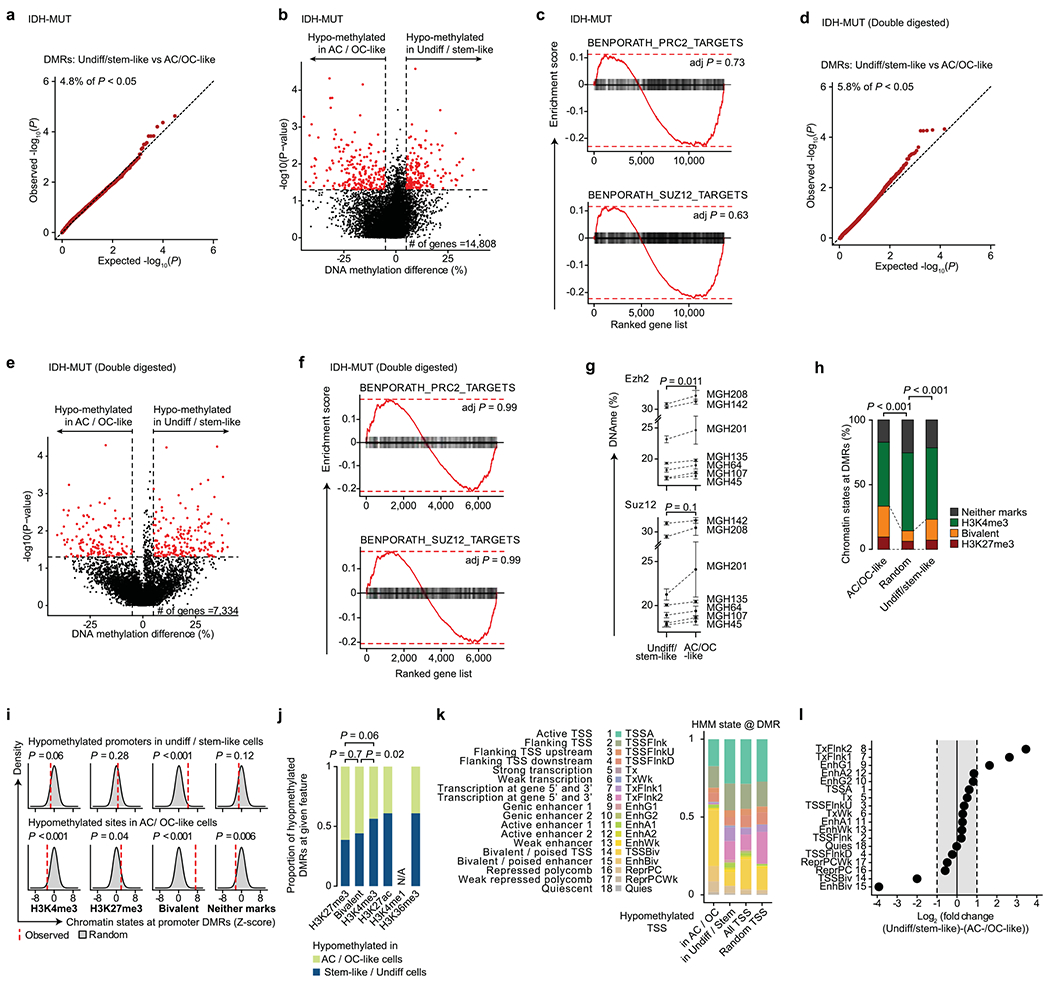
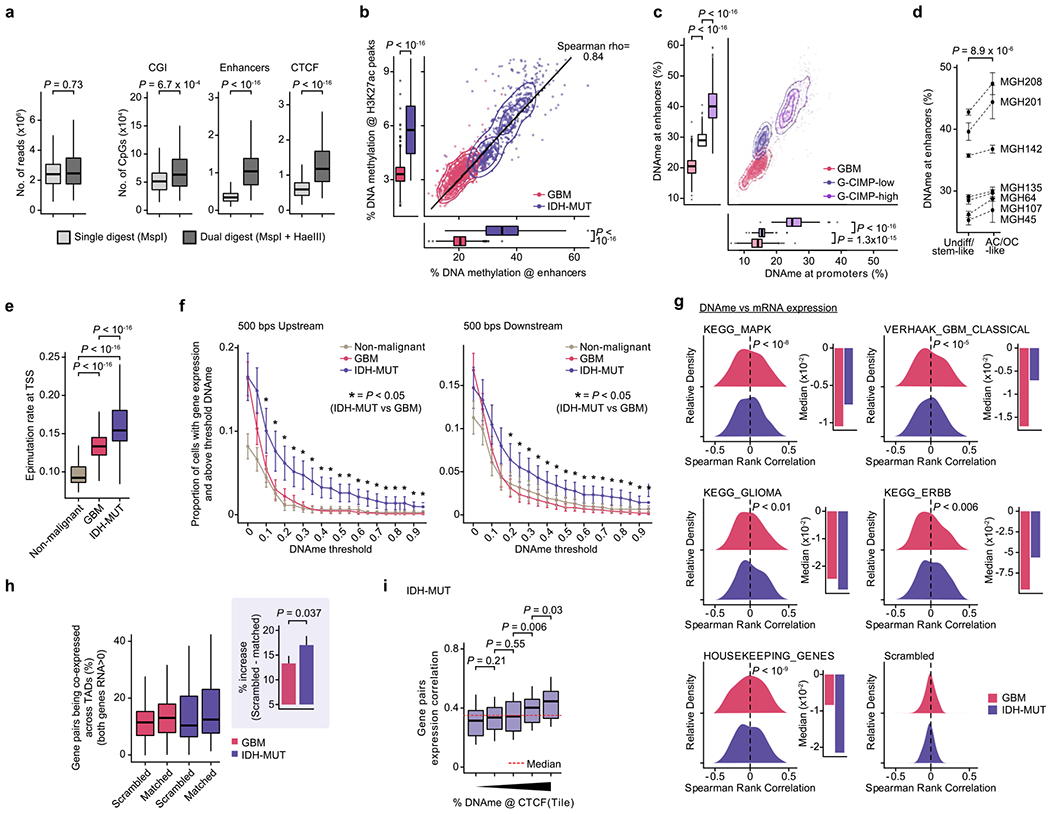
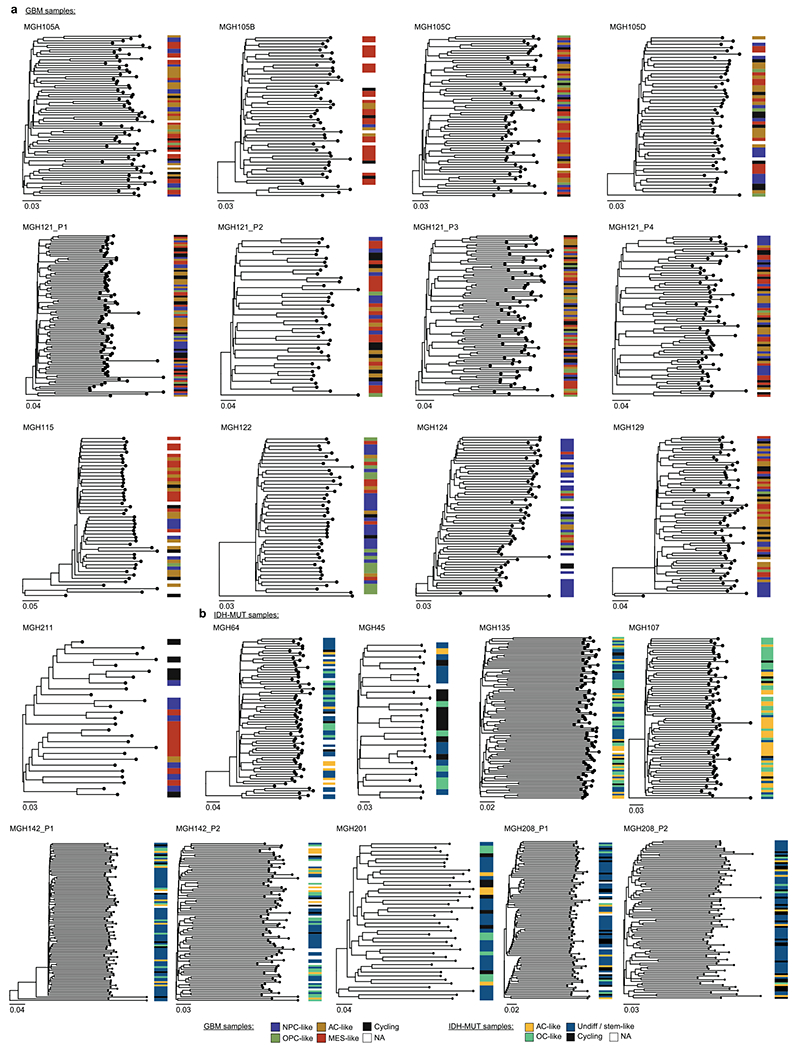
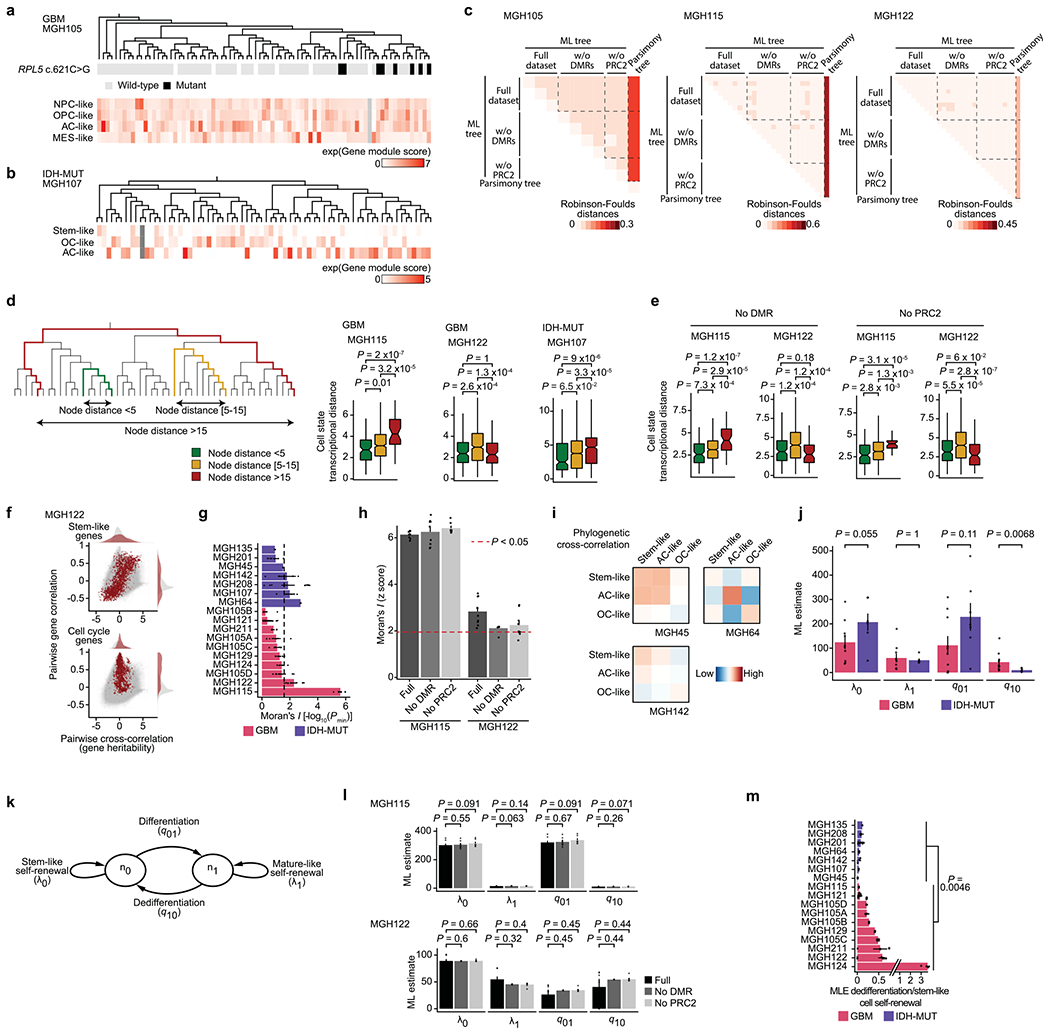
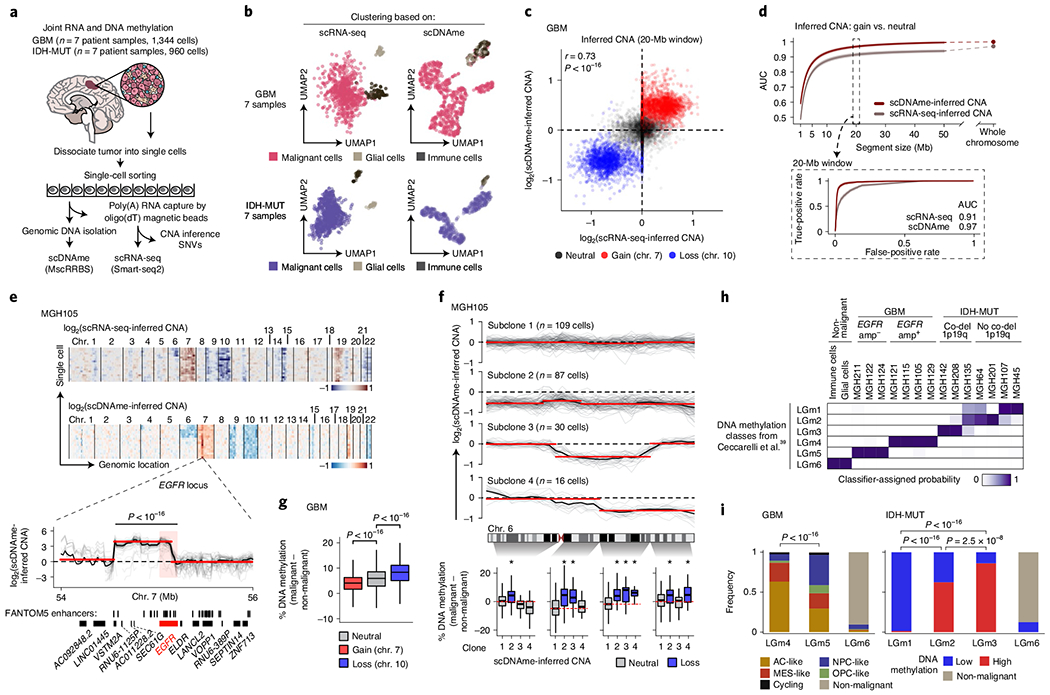
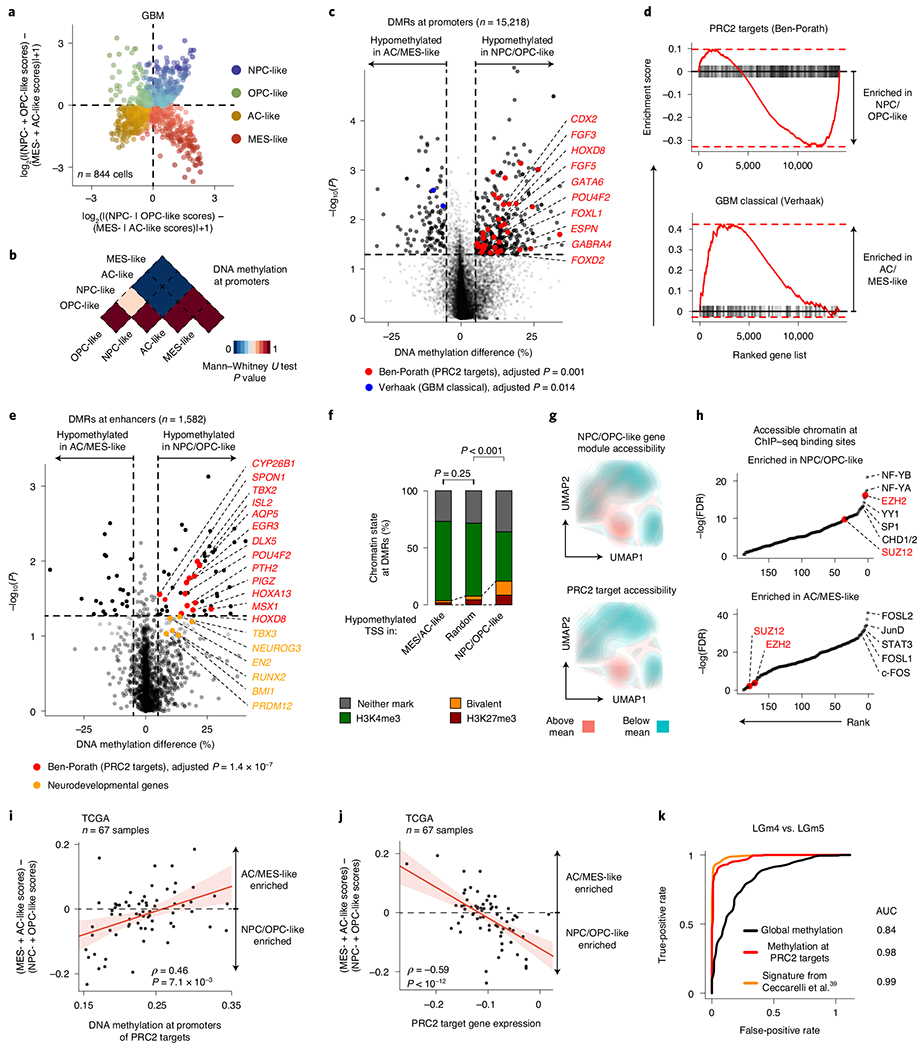
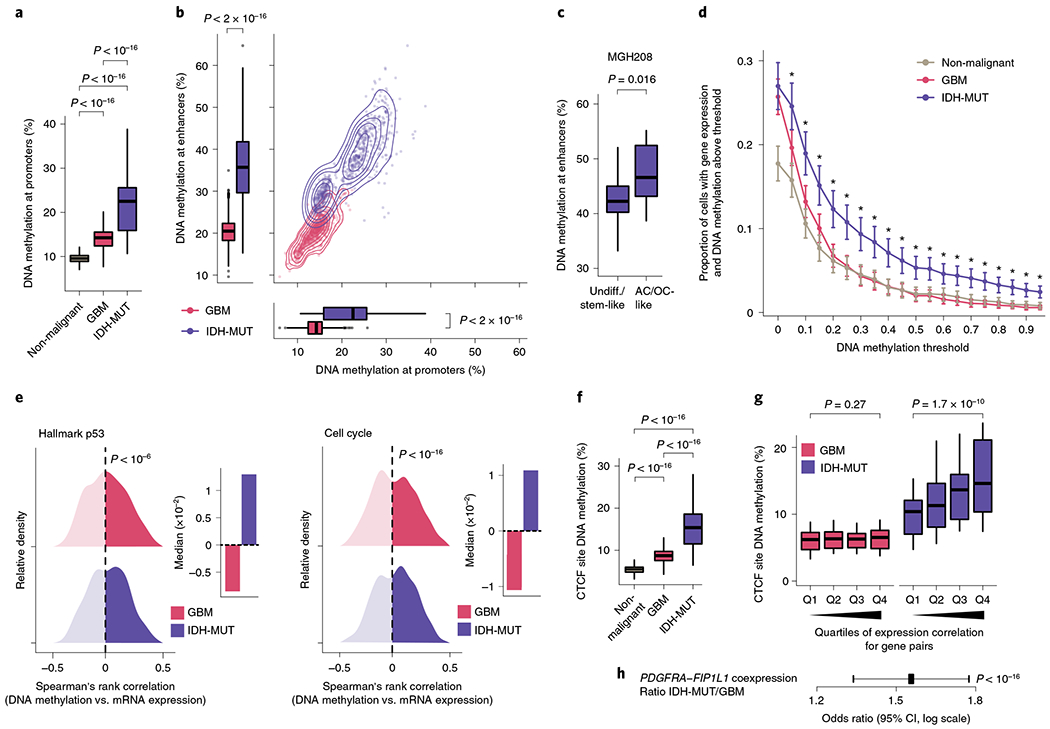
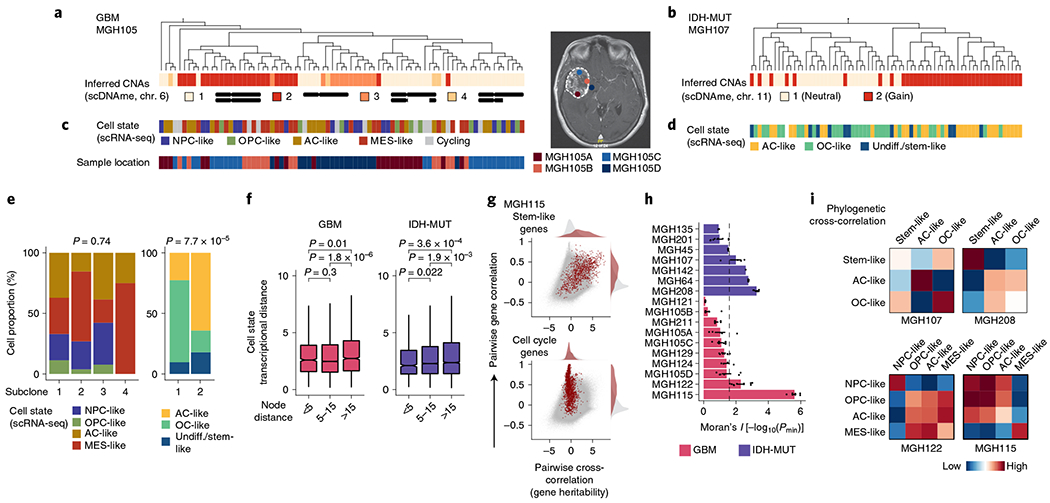
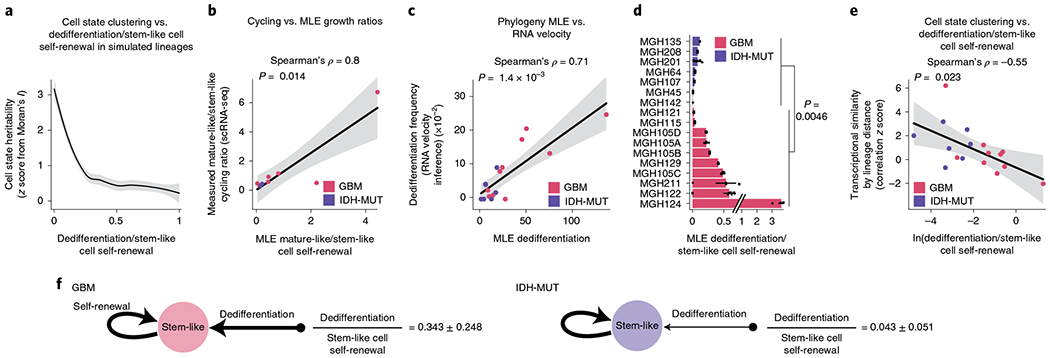
Single-cell RNA sequencing has revealed extensive transcriptional cell state diversity in cancer, often observed independently of genetic heterogeneity, raising the central question of how malignant cell states are encoded epigenetically. To address this, here we performed multiomics single-cell profiling-integrating DNA methylation, transcriptome and genotype within the same cells-of diffuse gliomas, tumors characterized by defined transcriptional cell state diversity. Direct comparison of the epigenetic profiles of distinct cell states revealed key switches for state transitions recapitulating neurodevelopmental trajectories and highlighted dysregulated epigenetic mechanisms underlying gliomagenesis. We further developed a quantitative framework to directly measure cell state heritability and transition dynamics based on high-resolution lineage trees in human samples. We demonstrated heritability of malignant cell states, with key differences in hierarchal and plastic cell state architectures in IDH-mutant glioma versus IDH-wild-type glioblastoma, respectively. This work provides a framework anchoring transcriptional cancer cell states in their epigenetic encoding, inheritance and transition dynamics.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
M.L.S. is an equity holder, scientific cofounder and advisory board member of Immunitas Therapeutics. A.R. is a founder and equity holder of Celsius Therapeutics, is an equity holder in Immunitas Therapeutics and, until 31 July 2020, was a scientific advisory board member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov and ThermoFisher Scientific. Since 1 August 2020, A.R. has been an employee of Genentech. Since 19 October 2020, O.R.-R. has been an employee of Genentech. D.A.L. is an equity holder, scientific cofounder and advisory board member of C2i Genomics and a scientific advisory board member for Mission Bio. The authors declare that these activities are not related to the research reported in this publication and have not influenced the conclusions in this manuscript. The remaining authors declare no competing interests.
Figures
Comment in
-
Epigenomic contributions to tumor cell heterogeneity and plasticity.Nat Genet. 2021 Oct;53(10):1403-1404. doi: 10.1038/s41588-021-00932-w. Nat Genet. 2021. PMID: 34608295 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
