

A cross-population atlas of genetic associations for 220 human phenotypes
- PMID: 34594039
- PMCID: PMC12208603
- DOI: 10.1038/s41588-021-00931-x
A cross-population atlas of genetic associations for 220 human phenotypes
Abstract
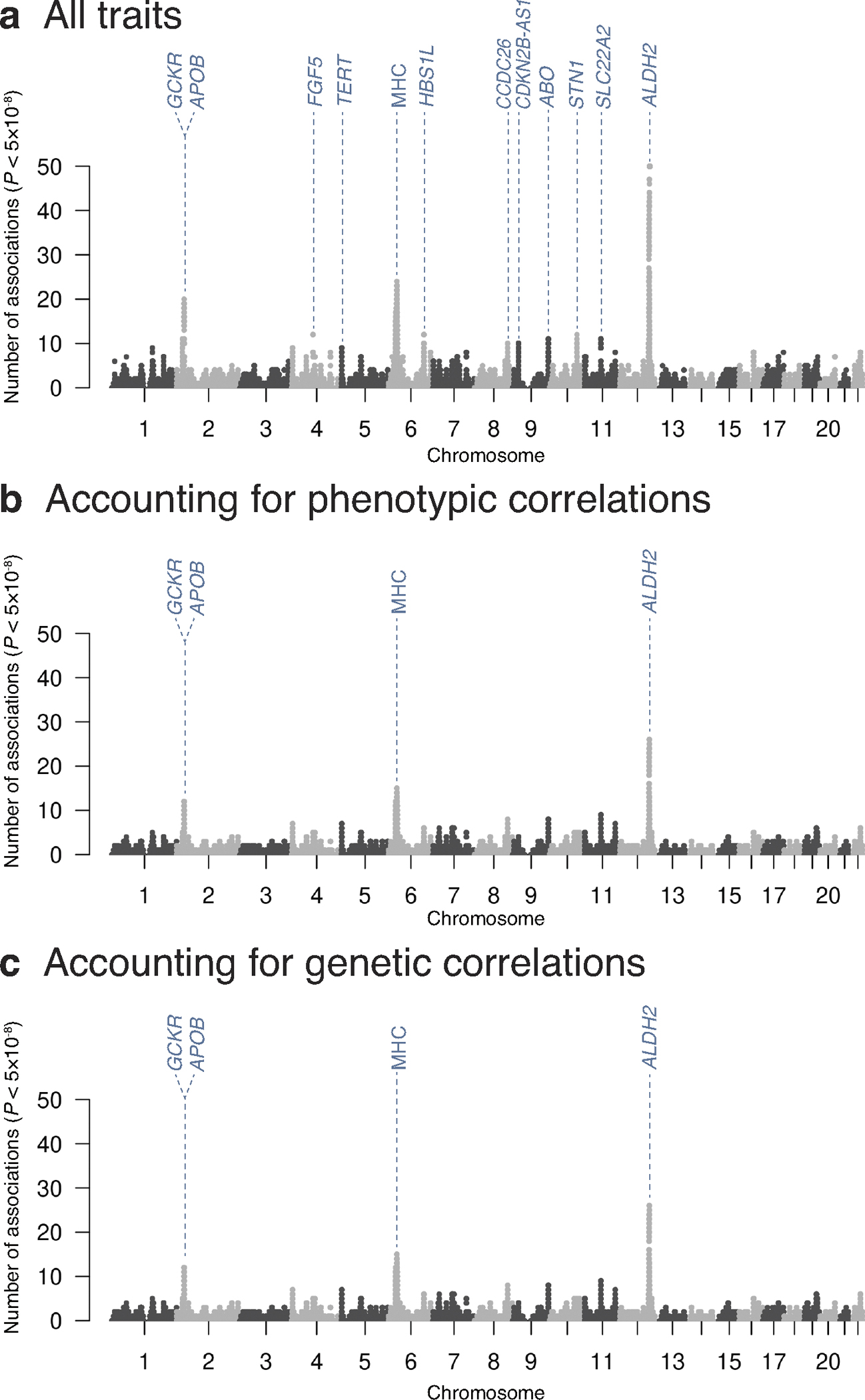
Current genome-wide association studies do not yet capture sufficient diversity in populations and scope of phenotypes. To expand an atlas of genetic associations in non-European populations, we conducted 220 deep-phenotype genome-wide association studies (diseases, biomarkers and medication usage) in BioBank Japan (n = 179,000), by incorporating past medical history and text-mining of electronic medical records. Meta-analyses with the UK Biobank and FinnGen (ntotal = 628,000) identified ~5,000 new loci, which improved the resolution of the genomic map of human traits. This atlas elucidated the landscape of pleiotropy as represented by the major histocompatibility complex locus, where we conducted HLA fine-mapping. Finally, we performed statistical decomposition of matrices of phenome-wide summary statistics, and identified latent genetic components, which pinpointed responsible variants and biological mechanisms underlying current disease classifications across populations. The decomposed components enabled genetically informed subtyping of similar diseases (for example, allergic diseases). Our study suggests a potential avenue for hypothesis-free re-investigation of human diseases through genetics.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Financial Interests
M.A.R. is on the SAB of 54Gene and Computational Advisory Board for Goldfinch Bio and has advised BioMarin, Third Rock Ventures, MazeTx and Related Sciences. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The remaining authors declare no competing interests.
Figures














References
-
- Berger D A brief history of medical diagnosis and the birth of the clinical laboratory. Part 1--Ancient times through the 19th century. MLO. Med. Lab. Obs. 31, (1999). - PubMed
-
- Organización Mundial de la Salud. International statistical classification of diseases and related health problems, 10th revision (ICD-10). World Heal. Organ. (2016).
Method Only References
-
- Sakaue S et al. Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan. Nat. Med. 26, 542–548 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
