

Lessons from Injury: How Nerve Injury Studies Reveal Basic Biological Mechanisms and Therapeutic Opportunities for Peripheral Nerve Diseases
- PMID: 34595734
- PMCID: PMC8804151
- DOI: 10.1007/s13311-021-01125-3
Lessons from Injury: How Nerve Injury Studies Reveal Basic Biological Mechanisms and Therapeutic Opportunities for Peripheral Nerve Diseases
Erratum in
-
Correction to: Lessons from Injury: How Nerve Injury Studies Reveal Basic Biological Mechanisms and Therapeutic Opportunities for Peripheral Nerve Diseases.Neurotherapeutics. 2021 Oct;18(4):2757. doi: 10.1007/s13311-021-01162-y. Neurotherapeutics. 2021. PMID: 34845709 Free PMC article. No abstract available.
Abstract
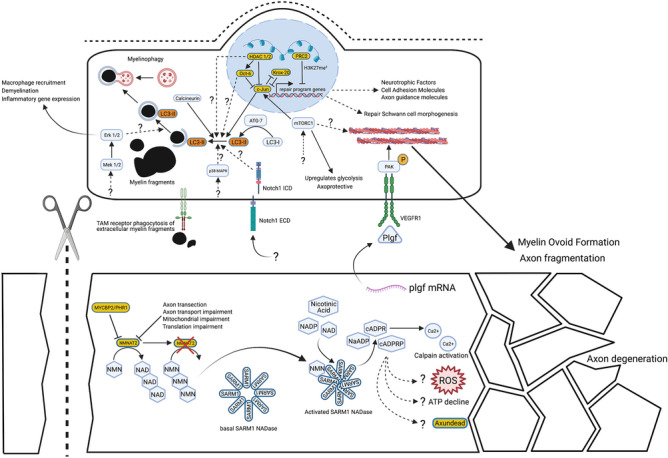
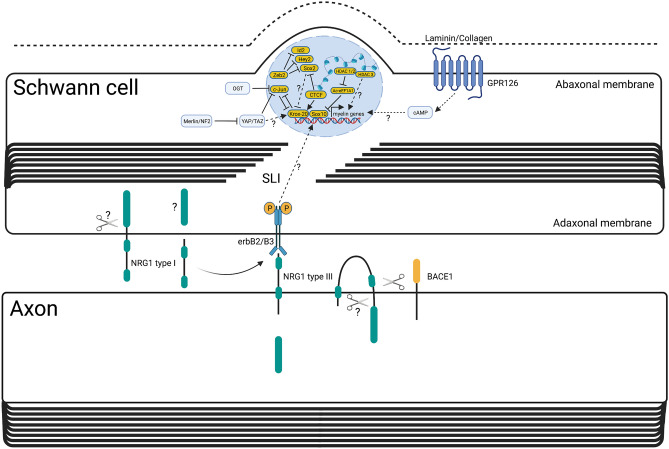
Since Waller and Cajal in the nineteenth and early twentieth centuries, laboratory traumatic peripheral nerve injury studies have provided great insight into cellular and molecular mechanisms governing axon degeneration and the responses of Schwann cells, the major glial cell type of peripheral nerves. It is now evident that pathways underlying injury-induced axon degeneration and the Schwann cell injury-specific state, the repair Schwann cell, are relevant to many inherited and acquired disorders of peripheral nerves. This review provides a timely update on the molecular understanding of axon degeneration and formation of the repair Schwann cell. We discuss how nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) and sterile alpha TIR motif containing protein 1 (SARM1) are required for axon survival and degeneration, respectively, how transcription factor c-JUN is essential for the Schwann cell response to nerve injury and what each tells us about disease mechanisms and potential therapies. Human genetic association with NMNAT2 and SARM1 strongly suggests aberrant activation of programmed axon death in polyneuropathies and motor neuron disorders, respectively, and animal studies suggest wider involvement including in chemotherapy-induced and diabetic neuropathies. In repair Schwann cells, cJUN is aberrantly expressed in a wide variety of human acquired and inherited neuropathies. Animal models suggest it limits axon loss in both genetic and traumatic neuropathies, whereas in contrast, Schwann cell secreted Neuregulin-1 type 1 drives onion bulb pathology in CMT1A. Finally, we discuss opportunities for drug-based and gene therapies to prevent axon loss or manipulate the repair Schwann cell state to treat acquired and inherited neuropathies and neuronopathies.
Keywords: C-JUN; NMNAT2; Neuregulin; Programmed axon death; Regeneration; Repair Schwann cell; SARM1; Wallerian degeneration.
© 2021. The Author(s).
Figures
References
-
- Ramon y Cajal, S. (1928). Degeneration and regeneration of the nervous system. Clarendon Press.
-
- Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primative fibres. Philosophical Transactions of the Royal Society of London. 1850;140:423–429. doi: 10.1098/rstl.1850.0021. - DOI
-
- Lubińska, L. (1982). Patterns of Wallerian degeneration of myelinated fibres in short and long peripheral stumps and in isolated segments of rat phrenic nerve. Interpretation of the role of axoplasmic flow of the trophic factor. Brain Research, 233(2), 227–240. 10.1016/0006-8993(82)91199-4 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
