

Advances in spatial transcriptomic data analysis
- PMID: 34599004
- PMCID: PMC8494229
- DOI: 10.1101/gr.275224.121
Advances in spatial transcriptomic data analysis
Abstract
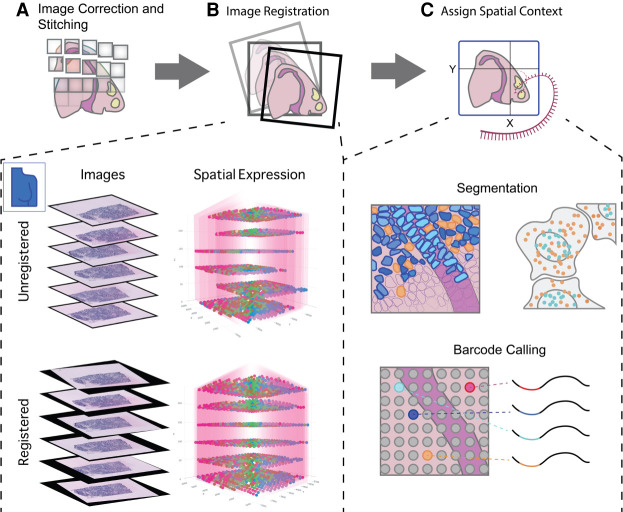
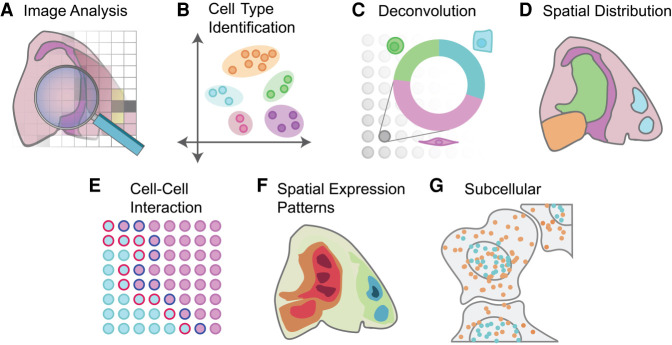
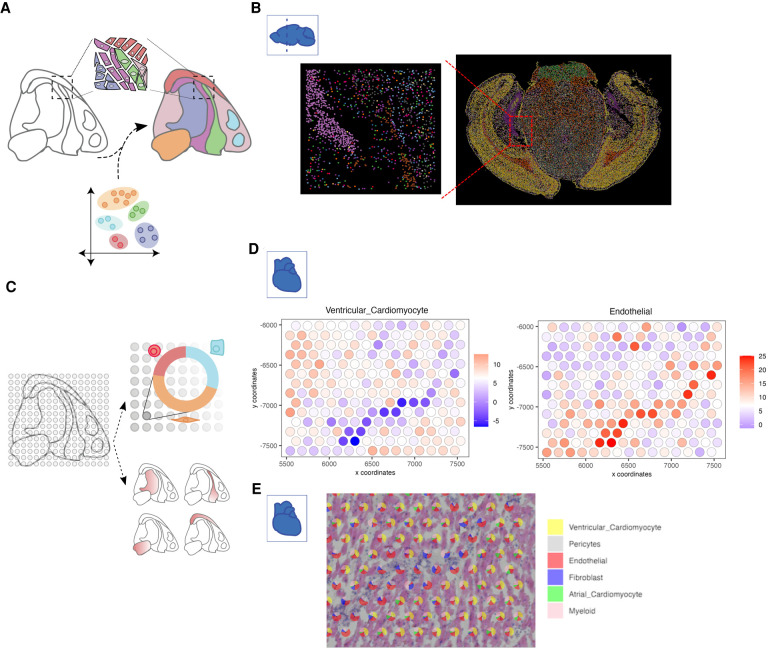
Spatial transcriptomics is a rapidly growing field that promises to comprehensively characterize tissue organization and architecture at the single-cell or subcellular resolution. Such information provides a solid foundation for mechanistic understanding of many biological processes in both health and disease that cannot be obtained by using traditional technologies. The development of computational methods plays important roles in extracting biological signals from raw data. Various approaches have been developed to overcome technology-specific limitations such as spatial resolution, gene coverage, sensitivity, and technical biases. Downstream analysis tools formulate spatial organization and cell-cell communications as quantifiable properties, and provide algorithms to derive such properties. Integrative pipelines further assemble multiple tools in one package, allowing biologists to conveniently analyze data from beginning to end. In this review, we summarize the state of the art of spatial transcriptomic data analysis methods and pipelines, and discuss how they operate on different technological platforms.
© 2021 Dries et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources