

239-kb Microdeletion Spanning KMT2E in a Child with Developmental Delay: Further Delineation of the Phenotype
- PMID: 34602960
- PMCID: PMC8436641
- DOI: 10.1159/000516635
239-kb Microdeletion Spanning KMT2E in a Child with Developmental Delay: Further Delineation of the Phenotype
Abstract
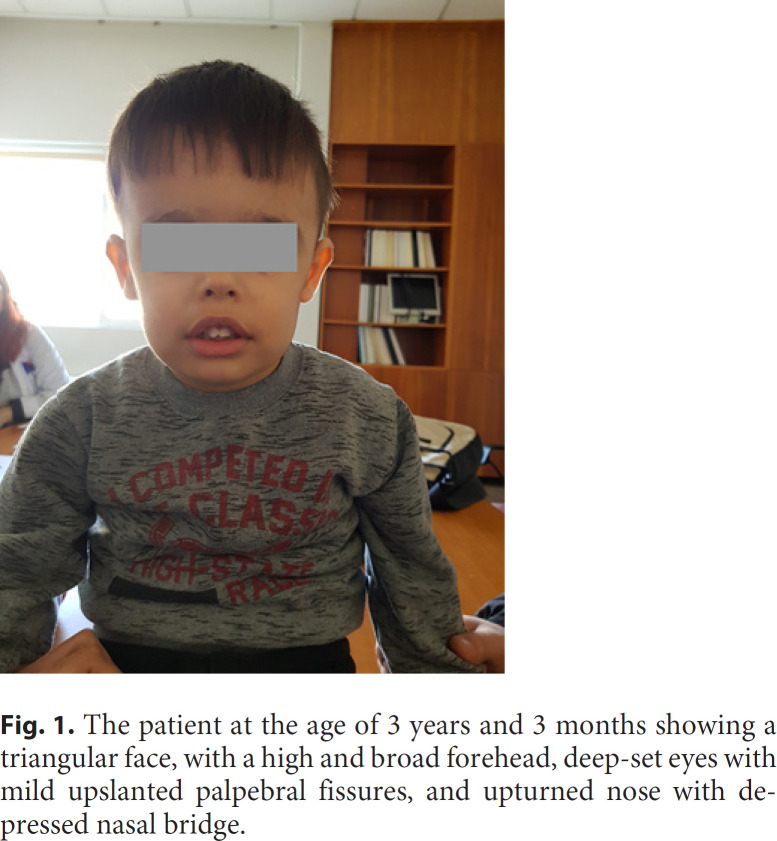
Pathogenic KMT2E variants underly O'Donnell-Luria-Rodan syndrome, a recently described neurodevelopmental disorder characterized by global developmental delay, variable degrees of intellectual disability, and subtle facial dysmorphism. Less common findings include autism, seizures, gastrointestinal (GI) problems, and abnormal head circumference. Occurrence of mostly truncating variants as well as the similar phenotype observed in individuals with deletions spanning KMT2E suggest haploinsufficiency of this gene as a common mechanism for the disorder, while a gain-of-function or dominant-negative effect cannot be ruled out for some missense variants. Deletions reported in the literature encompass several additional known or presumed haploinsufficient genes, thus leading to more complex phenotypes. Here, we describe a male with antenatal onset hydronephrosis, hypotonia, global developmental delay, prominent GI symptoms as well as facial dysmorphism. Chromosomal microarray revealed a 239-kb de novo microdeletion spanning KMT2E and LHFPL3. Clinical presentation of our proband, harboring one of the smallest deletions of the region confirms the core features of this disorder, suggests GI symptoms as a prominent finding in affected individuals while expanding the phenotypic spectrum to abnormalities of the urinary tract.
Keywords: 7q22.3 microdeletion; Developmental delay; KMT2E; LHFPL3; O'Donnell-Luria-Rodan syndrome.
Copyright © 2021 by S. Karger AG, Basel.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures

Similar articles
-
Case Report: De novo Variants of KMT2E Cause O'Donnell-Luria-Rodan Syndrome: Additional Cases and Literature Review.Front Pediatr. 2021 Feb 18;9:641841. doi: 10.3389/fped.2021.641841. eCollection 2021. Front Pediatr. 2021. PMID: 33681112 Free PMC article.
-
Molecular and clinical Insights into KMT2E-Related O'Donnell-Luria-Rodan syndrome in a novel patient cohort.Eur J Med Genet. 2025 Feb;73:104990. doi: 10.1016/j.ejmg.2024.104990. Epub 2024 Dec 19. Eur J Med Genet. 2025. PMID: 39709003
-
KMT2E-Related Neurodevelopmental Disorder.2024 Apr 18. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2024 Apr 18. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 38648332 Free Books & Documents. Review.
-
A Novel KMT2E Splicing Variant as a Cause of O'Donnell-Luria-Rodan Syndrome With West Syndrome: Expansion of the Phenotype and Genotype.Int J Dev Neurosci. 2025 Apr;85(2):e70012. doi: 10.1002/jdn.70012. Int J Dev Neurosci. 2025. PMID: 40070083
-
ODLURO syndrome: personal experience and review of the literature.Radiol Med. 2021 Feb;126(2):316-322. doi: 10.1007/s11547-020-01255-2. Epub 2020 Jul 20. Radiol Med. 2021. PMID: 32691224 Review.
Cited by
-
Clinical and genetic analysis of a case of O'Donnell-Luria-Rodan syndrome manifesting as growth retardation.Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2024 Apr 28;49(4):649-654. doi: 10.11817/j.issn.1672-7347.2024.230359. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2024. PMID: 39019795 Free PMC article. Chinese, English.
References
-
- Conforti R, Iovine S, Santangelo G, Capasso R, Cirillo M, Fratta M, et al. ODLURO syndrome: personal experience and review of the literature. Radiol Med. 2021;126((2)):316–22. - PubMed
-
- Damen-Elias HA, De Jong TP, Stigter RH, Visser GH, Stoutenbeek PH. Congenital renal tract anomalies: outcome and follow-up of 402 cases detected antenatally between 1986 and 2001. Ultrasound Obstet Gynecol. 2005;25:134–43. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
