

Innate Immune System Activation and Neuroinflammation in Down Syndrome and Neurodegeneration: Therapeutic Targets or Partners?
- PMID: 34603007
- PMCID: PMC8481947
- DOI: 10.3389/fnagi.2021.718426
Innate Immune System Activation and Neuroinflammation in Down Syndrome and Neurodegeneration: Therapeutic Targets or Partners?
Abstract
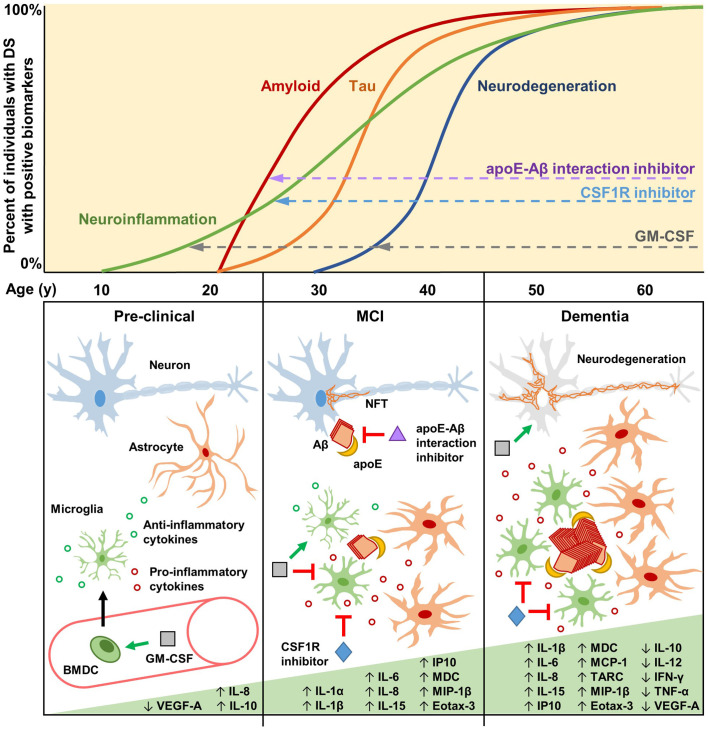
Innate immune system activation and inflammation are associated with and may contribute to clinical outcomes in people with Down syndrome (DS), neurodegenerative diseases such as Alzheimer's disease (AD), and normal aging. In addition to serving as potential diagnostic biomarkers, innate immune system activation and inflammation may play a contributing or causal role in these conditions, leading to the hypothesis that effective therapies should seek to dampen their effects. However, recent intervention studies with the innate immune system activator granulocyte-macrophage colony-stimulating factor (GM-CSF) in animal models of DS, AD, and normal aging, and in an AD clinical trial suggest that activating the innate immune system and inflammation may instead be therapeutic. We consider evidence that DS, AD, and normal aging are accompanied by innate immune system activation and inflammation and discuss whether and when during the disease process it may be therapeutically beneficial to suppress or promote such activation.
Keywords: Alzheimer’s disease; Down syndrome; GM-CSF (granulocyte-macrophage colony-stimulating factor); amyloid-β; apolipoprotein E; drug repurposing and discovery; inflammation; innate immune system.
Copyright © 2021 Ahmed, Johnson, Boyd, Coughlan, Chial and Potter.
Conflict of interest statement
HP and TB are two of the inventors on several U.S. patents owned by the University of South Florida, but not licensed. TB is an employee of Partner Therapeutics. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
The innate immune system stimulating cytokine GM-CSF improves learning/memory and interneuron and astrocyte brain pathology in Dp16 Down syndrome mice and improves learning/memory in wild-type mice.Neurobiol Dis. 2022 Jun 15;168:105694. doi: 10.1016/j.nbd.2022.105694. Epub 2022 Mar 18. Neurobiol Dis. 2022. PMID: 35307513 Free PMC article.
-
Safety and efficacy of sargramostim (GM-CSF) in the treatment of Alzheimer's disease.Alzheimers Dement (N Y). 2021 Mar 24;7(1):e12158. doi: 10.1002/trc2.12158. eCollection 2021. Alzheimers Dement (N Y). 2021. PMID: 33778150 Free PMC article.
-
Granulocyte macrophage colony-stimulating factor and the intestinal innate immune cell homeostasis in Crohn's disease.Am J Physiol Gastrointest Liver Physiol. 2014 Mar;306(6):G455-65. doi: 10.1152/ajpgi.00409.2013. Epub 2014 Feb 6. Am J Physiol Gastrointest Liver Physiol. 2014. PMID: 24503766 Review.
-
The antimicrobial protection hypothesis of Alzheimer's disease.Alzheimers Dement. 2018 Dec;14(12):1602-1614. doi: 10.1016/j.jalz.2018.06.3040. Epub 2018 Oct 9. Alzheimers Dement. 2018. PMID: 30314800
-
Innate neuroimmunity across aging and neurodegeneration: a perspective from amyloidogenic evolvability.Front Cell Dev Biol. 2024 Jul 12;12:1430593. doi: 10.3389/fcell.2024.1430593. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39071802 Free PMC article. Review.
Cited by
-
Reversing intellectual disabilities in Down syndrome: Hopes or hypes?Indian J Med Res. 2025 Mar;161(3):203-206. doi: 10.25259/IJMR_646_2025. Indian J Med Res. 2025. PMID: 40347506 Free PMC article. No abstract available.
-
Suppressor of Cytokine Signalling 5 (SOCS5) Modulates Inflammatory Responses during Alphavirus Infection.Viruses. 2022 Nov 9;14(11):2476. doi: 10.3390/v14112476. Viruses. 2022. PMID: 36366574 Free PMC article.
-
Risk of Alzheimer's disease in Down syndrome: Insights gained by multi-omics.Alzheimers Dement. 2025 Apr;21(4):e14604. doi: 10.1002/alz.14604. Alzheimers Dement. 2025. PMID: 40207399 Free PMC article. Review.
-
Alzheimer's disease is an inherent, natural part of human brain aging: an integrated perspective.Free Neuropathol. 2022 Jul 8;3:17. doi: 10.17879/freeneuropathology-2022-3806. eCollection 2022 Jan. Free Neuropathol. 2022. PMID: 37284149 Free PMC article.
-
Cell type characterization of spatiotemporal gene co-expression modules in Down syndrome brain.iScience. 2022 Dec 28;26(1):105884. doi: 10.1016/j.isci.2022.105884. eCollection 2023 Jan 20. iScience. 2022. PMID: 36647384 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources