

Loop Diuretics Inhibit Ischemia-Induced Intracellular Ca2+ Overload in Neurons via the Inhibition of Voltage-Gated Ca2+ and Na+ Channels
- PMID: 34603048
- PMCID: PMC8479115
- DOI: 10.3389/fphar.2021.732922
Loop Diuretics Inhibit Ischemia-Induced Intracellular Ca2+ Overload in Neurons via the Inhibition of Voltage-Gated Ca2+ and Na+ Channels
Abstract
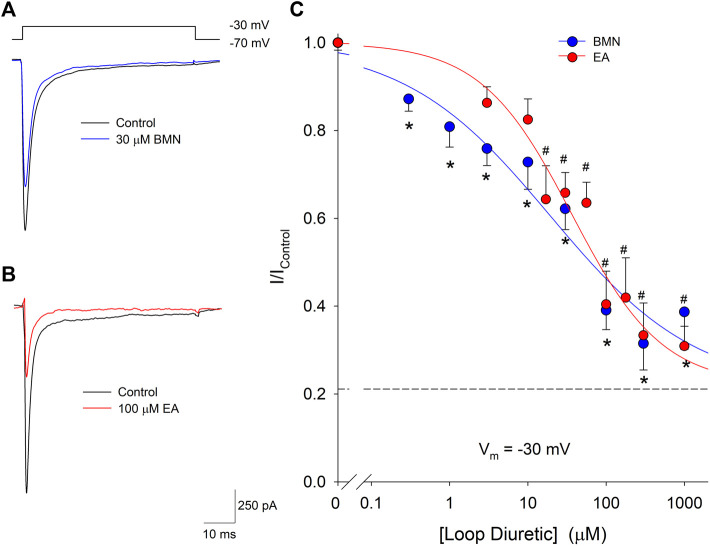
One consequence of ischemic stroke is disruption of intracellular ionic homeostasis. Intracellular overload of both Na+ and Ca2+ has been linked to neuronal death in this pathophysiological state. The etiology of ionic imbalances resulting from stroke-induced ischemia and acidosis includes the dysregulation of multiple plasma membrane transport proteins, such as increased activity of sodium-potassium-chloride cotransporter-1 (NKCC-1). Experiments using NKCC1 antagonists, bumetanide (BMN) and ethacrynic acid (EA), were carried out to determine if inhibition of this cotransporter affects Na+ and Ca2+ overload observed following in vitro ischemia-acidosis. Fluorometric Ca2+ and Na+ measurements were performed using cultured cortical neurons, and measurements of whole-cell membrane currents were used to determine target(s) of BMN and EA, other than the electroneutral NKCC-1. Both BMN and EA depressed ischemia-acidosis induced [Ca2+]i overload without appreciably reducing [Na+]i increases. Voltage-gated Ca2+ channels were inhibited by both BMN and EA with half-maximal inhibitory concentration (IC50) values of 4 and 36 μM, respectively. Similarly, voltage-gated Na+ channels were blocked by BMN and EA with IC50 values of 13 and 30 μM, respectively. However, neither BMN nor EA affected currents mediated by acid-sensing ion channels or ionotropic glutamatergic receptors, both of which are known to produce [Ca2+]i overload following ischemia. Data suggest that loop diuretics effectively inhibit voltage-gated Ca2+ and Na+ channels at clinically relevant concentrations, and block of these channels by these compounds likely contributes to their clinical effects. Importantly, inhibition of these channels, and not NKCC1, by loop diuretics reduces [Ca2+]i overload in neurons during ischemia-acidosis, and thus BMN and EA could potentially be used therapeutically to lessen injury following ischemic stroke.
Keywords: acidosis; bumetanide; calcium; ethacrynic acid; ischemia; neurons; sodium; voltage-gated channels.
Copyright © 2021 Katnik and Cuevas.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures






Similar articles
-
ASIC1a channels are activated by endogenous protons during ischemia and contribute to synergistic potentiation of intracellular Ca(2+) overload during ischemia and acidosis.Cell Calcium. 2010 Jul;48(1):70-82. doi: 10.1016/j.ceca.2010.07.002. Epub 2010 Aug 1. Cell Calcium. 2010. PMID: 20678793
-
Afobazole modulates neuronal response to ischemia and acidosis via activation of sigma-1 receptors.J Pharmacol Exp Ther. 2011 Oct;339(1):152-60. doi: 10.1124/jpet.111.182774. Epub 2011 Jun 29. J Pharmacol Exp Ther. 2011. PMID: 21715562
-
Extracellular acidosis contracts coronary but neither renal nor mesenteric artery via modulation of H+,K+-ATPase, voltage-gated K+ channels and L-type Ca2+ channels.Exp Physiol. 2014 Jul;99(7):995-1006. doi: 10.1113/expphysiol.2014.078634. Epub 2014 May 16. Exp Physiol. 2014. PMID: 24928954
-
The role of Na-K-Cl co-transporter in cerebral ischemia.Neurol Res. 2005 Apr;27(3):280-6. doi: 10.1179/016164105X25243. Neurol Res. 2005. PMID: 15845211 Review.
-
Ion channels and transporters in microglial function in physiology and brain diseases.Neurochem Int. 2021 Jan;142:104925. doi: 10.1016/j.neuint.2020.104925. Epub 2020 Nov 26. Neurochem Int. 2021. PMID: 33248207 Free PMC article. Review.
Cited by
-
Ancient Chinese Herbal Recipe Huanglian Jie Du Decoction for Ischemic Stroke: An Overview of Current Evidence.Aging Dis. 2022 Dec 1;13(6):1733-1744. doi: 10.14336/AD.2022.0311. eCollection 2022 Dec 1. Aging Dis. 2022. PMID: 36465168 Free PMC article.
-
Ethacrynic Acid: A Promising Candidate for Drug Repurposing as an Anticancer Agent.Int J Mol Sci. 2023 Apr 4;24(7):6712. doi: 10.3390/ijms24076712. Int J Mol Sci. 2023. PMID: 37047688 Free PMC article. Review.
References
-
- Chen X., Kintner D. B., Luo J., Baba A., Matsuda T., Sun D. (2008). Endoplasmic Reticulum Ca2+ Dysregulation and Endoplasmic Reticulum Stress Following In Vitro Neuronal Ischemia: Role of Na+-K+-Cl- Cotransporter. J. Neurochem. 106 (4), 1563–1576. 10.1111/j.1471-4159.2008.05501.x - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous