

Serotyping, MLST, and Core Genome MLST Analysis of Salmonella enterica From Different Sources in China During 2004-2019
- PMID: 34603224
- PMCID: PMC8481815
- DOI: 10.3389/fmicb.2021.688614
Serotyping, MLST, and Core Genome MLST Analysis of Salmonella enterica From Different Sources in China During 2004-2019
Abstract
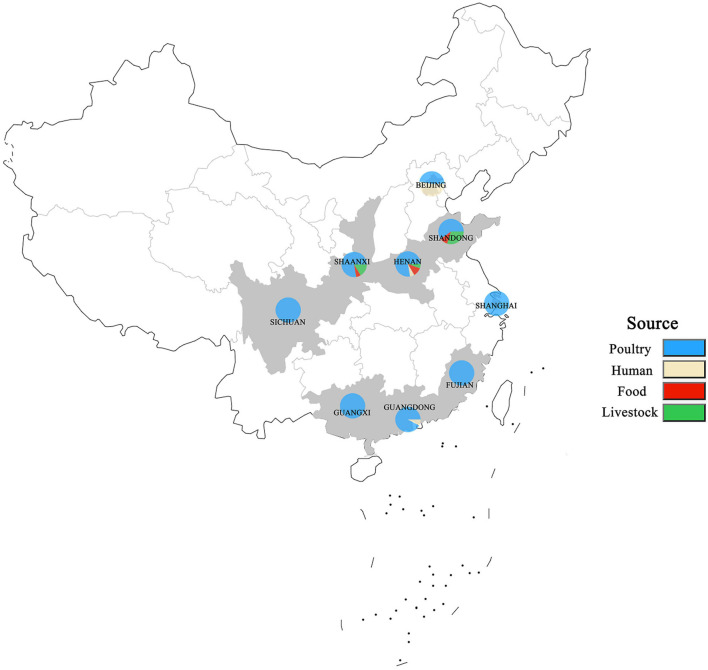
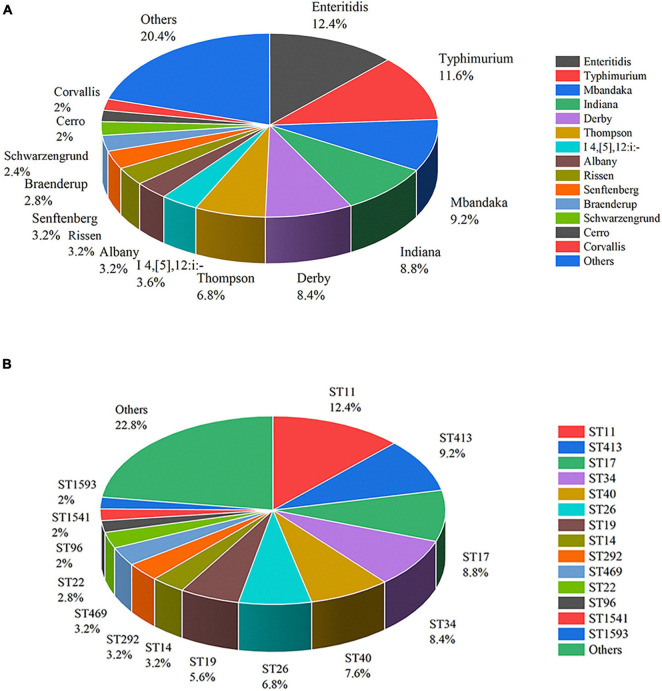
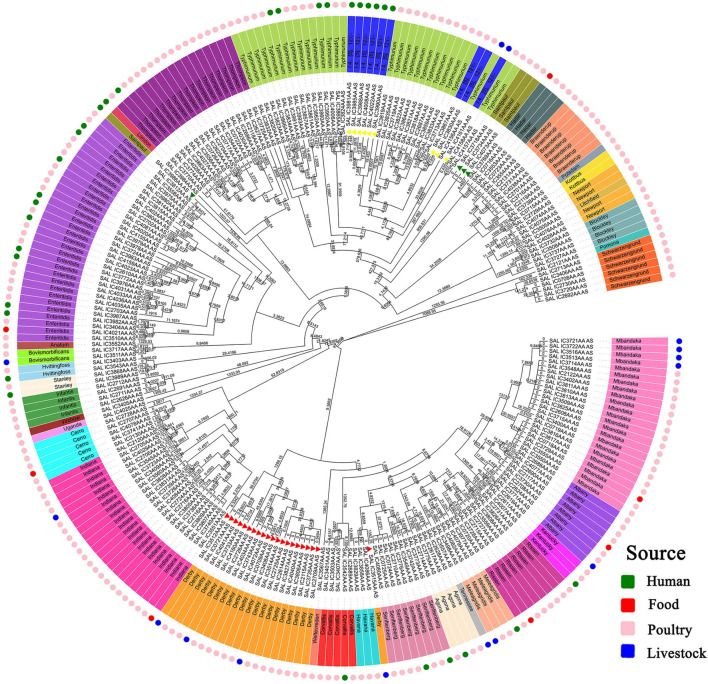
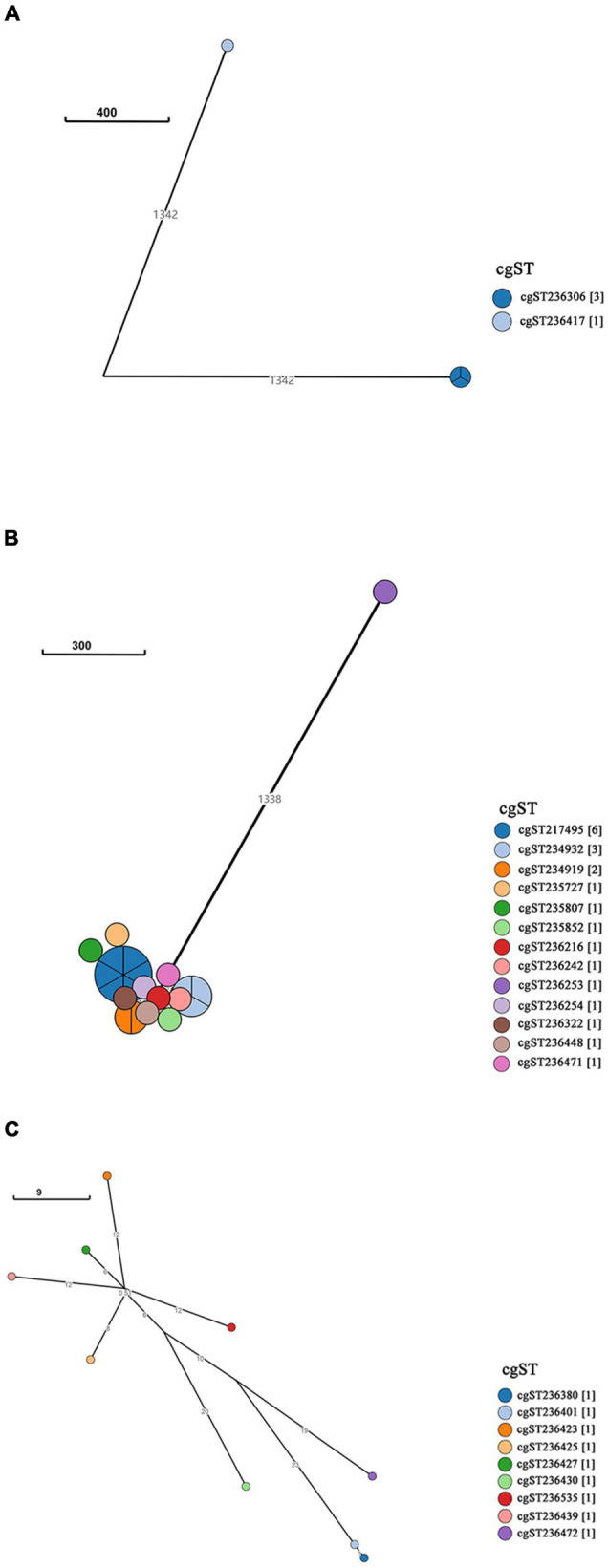
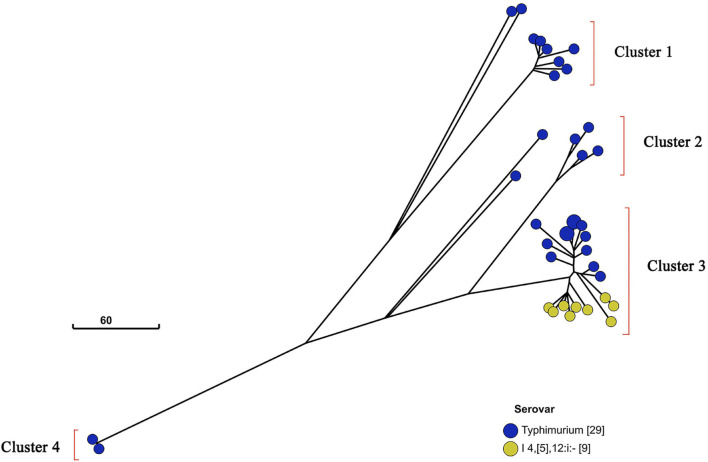
Salmonella enterica (S. enterica) is an important foodborne pathogen, causing food poisoning and human infection, and critically threatening food safety and public health. Salmonella typing is essential for bacterial identification, tracing, epidemiological investigation, and monitoring. Serotyping and multilocus sequence typing (MLST) analysis are standard bacterial typing methods despite the low resolution. Core genome MLST (cgMLST) is a high-resolution molecular typing method based on whole genomic sequencing for accurate bacterial tracing. We investigated 250 S. enterica isolates from poultry, livestock, food, and human sources in nine provinces of China from 2004 to 2019 using serotyping, MLST, and cgMLST analysis. All S. enterica isolates were divided into 36 serovars using slide agglutination. The major serovars in order were Enteritidis (31 isolates), Typhimurium (29 isolates), Mbandaka (23 isolates), and Indiana (22 isolates). All strains were assigned into 43 sequence types (STs) by MLST. Among them, ST11 (31 isolates) was the primary ST. Besides this, a novel ST, ST8016, was identified, and it was different from ST40 by position 317 C → T in dnaN. Furthermore, these 250 isolates were grouped into 185 cgMLST sequence types (cgSTs) by cgMLST. The major cgST was cgST235530 (11 isolates), and only three cgSTs contained isolates from human and other sources, indicating a possibility of cross-species infection. Phylogenetic analysis indicated that most of the same serovar strains were putatively homologous except Saintpaul and Derby due to their multilineage characteristics. In addition, serovar I 4,[5],12:i:- and Typhimurium isolates have similar genomic relatedness on the phylogenetic tree. In conclusion, we sorted out the phenotyping and genotyping diversity of S. enterica isolates in China during 2004-2019 and clarified the temporal and spatial distribution characteristics of Salmonella from different hosts in China in the recent 16 years. These results greatly supplement Salmonella strain resources, genetic information, and traceability typing data; facilitate the typing, traceability, identification, and genetic evolution analysis of Salmonella; and therefore, improve the level of analysis, monitoring, and controlling of foodborne microorganisms in China.
Keywords: MLST; Salmonella enterica; cgMLST; serotype; whole genome sequencing.
Copyright © 2021 Yan, Zhang, Li, Liu, Zhu, Chen and Yang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Genomic typing and virulence gene profile analysis of Salmonella Derby from different sources.Microb Pathog. 2023 Sep;182:106248. doi: 10.1016/j.micpath.2023.106248. Epub 2023 Jul 7. Microb Pathog. 2023. PMID: 37423493
-
Genomes-based MLST, cgMLST, wgMLST and SNP analysis of Salmonella Typhimurium from animals and humans.Comp Immunol Microbiol Infect Dis. 2023 May;96:101973. doi: 10.1016/j.cimid.2023.101973. Epub 2023 Mar 23. Comp Immunol Microbiol Infect Dis. 2023. PMID: 36989679
-
Comparative analysis of core genome MLST and SNP typing within a European Salmonella serovar Enteritidis outbreak.Int J Food Microbiol. 2018 Jun 2;274:1-11. doi: 10.1016/j.ijfoodmicro.2018.02.023. Epub 2018 Feb 28. Int J Food Microbiol. 2018. PMID: 29574242 Free PMC article.
-
Molecular methods for serovar determination of Salmonella.Crit Rev Microbiol. 2015;41(3):309-25. doi: 10.3109/1040841X.2013.837862. Epub 2013 Nov 14. Crit Rev Microbiol. 2015. PMID: 24228625 Review.
-
A genomic overview of the population structure of Salmonella.PLoS Genet. 2018 Apr 5;14(4):e1007261. doi: 10.1371/journal.pgen.1007261. eCollection 2018 Apr. PLoS Genet. 2018. PMID: 29621240 Free PMC article. Review.
Cited by
-
Plasmid Composition, Antimicrobial Resistance and Virulence Genes Profiles of Ciprofloxacin- and Third-Generation Cephalosporin-Resistant Foodborne Salmonella enterica Isolates from Russia.Microorganisms. 2023 Jan 30;11(2):347. doi: 10.3390/microorganisms11020347. Microorganisms. 2023. PMID: 36838312 Free PMC article.
-
The Resistance and Virulence Characteristics of Salmonella Enteritidis Strain Isolated from Patients with Food Poisoning Based on the Whole-Genome Sequencing and Quantitative Proteomic Analysis.Infect Drug Resist. 2023 Oct 6;16:6567-6586. doi: 10.2147/IDR.S411125. eCollection 2023. Infect Drug Resist. 2023. PMID: 37823028 Free PMC article.
-
Epidemic trend of Salmonella from swines and broilers in China from 2014 to 2023 and genetic evolution analysis of ESBLs-producing strains.Front Microbiol. 2025 Feb 14;16:1510751. doi: 10.3389/fmicb.2025.1510751. eCollection 2025. Front Microbiol. 2025. PMID: 40028455 Free PMC article.
-
In vitro and in silico parameters for precise cgMLST typing of Listeria monocytogenes.BMC Genomics. 2022 Mar 26;23(1):235. doi: 10.1186/s12864-022-08437-4. BMC Genomics. 2022. PMID: 35346021 Free PMC article.
-
Genomic and proteomic analysis of Salmonella Enteritidis isolated from a patient with foodborne diarrhea.World J Microbiol Biotechnol. 2023 Dec 20;40(2):48. doi: 10.1007/s11274-023-03857-0. World J Microbiol Biotechnol. 2023. PMID: 38114804
References
-
- Almeida F., Amanda A. S., Marta I. C. M., Dália dos P. R., Alessandro De M. V., Yan L., et al. (2018). Phylogenetic and antimicrobial resistance gene analysis of Salmonella Typhimurium strains isolated in Brazil by whole genome sequencing. PLoS One 13:e0201882. 10.1371/journal.pone.0201882 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources