

Nuclear Mechanisms Involved in Endocrine Resistance
- PMID: 34604071
- PMCID: PMC8480308
- DOI: 10.3389/fonc.2021.736597
Nuclear Mechanisms Involved in Endocrine Resistance
Abstract
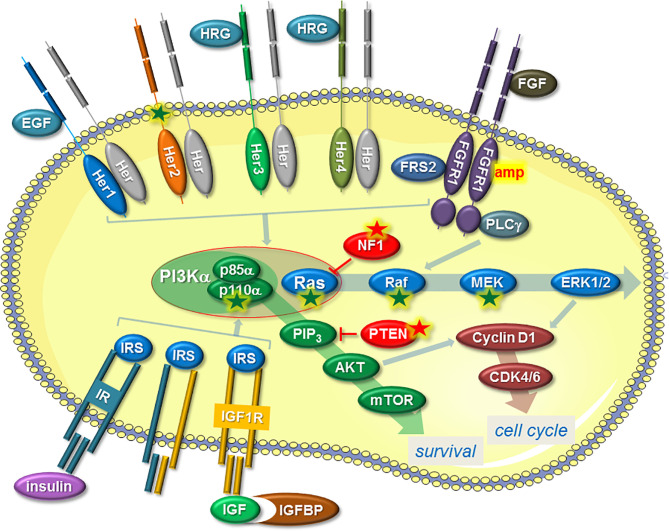
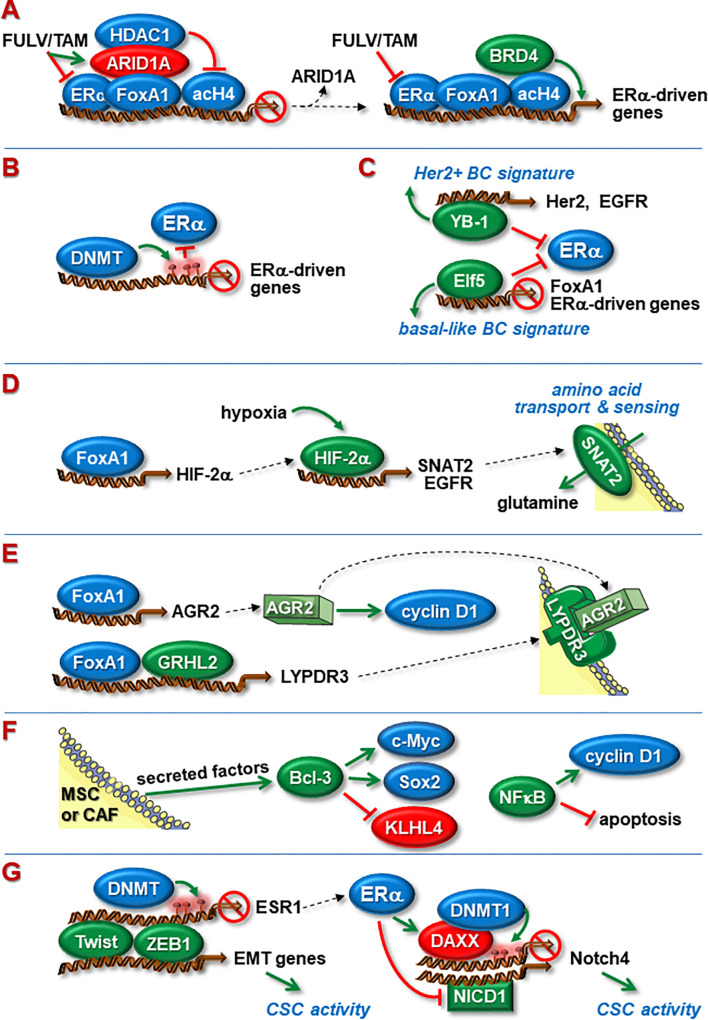
Endocrine therapy is a standard treatment offered to patients with ERα (estrogen receptor α)-positive breast cancer. In endocrine therapy, ERα is either directly targeted by anti-estrogens or indirectly by aromatase inhibitors which cause estrogen deficiency. Resistance to these drugs (endocrine resistance) compromises the efficiency of this treatment and requires additional measures. Endocrine resistance is often caused by deregulation of the PI3K/AKT/mTOR pathway and/or cyclin-dependent kinase 4 and 6 activities allowing inhibitors of these factors to be used clinically to counteract endocrine resistance. The nuclear mechanisms involved in endocrine resistance are beginning to emerge. Exploring these mechanisms may reveal additional druggable targets, which could help to further improve patients' outcome in an endocrine resistance setting. This review intends to summarize our current knowledge on the nuclear mechanisms linked to endocrine resistance.
Keywords: cancer stem cells; chromatin accessibility; estrogen receptor; fulvestrant; tamoxifen; transcription factors; transcriptional reprogramming.
Copyright © 2021 Dittmer.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous