

Markov State Models to Study the Functional Dynamics of Proteins in the Wake of Machine Learning
- PMID: 34604842
- PMCID: PMC8479766
- DOI: 10.1021/jacsau.1c00254
Markov State Models to Study the Functional Dynamics of Proteins in the Wake of Machine Learning
Abstract
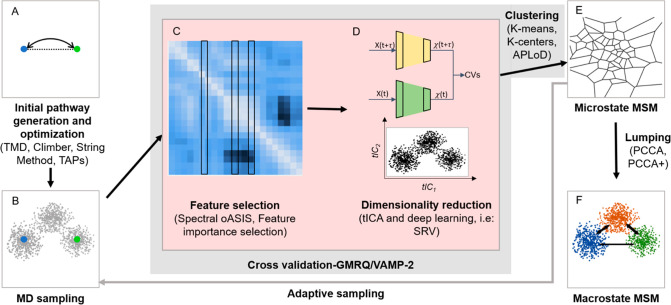
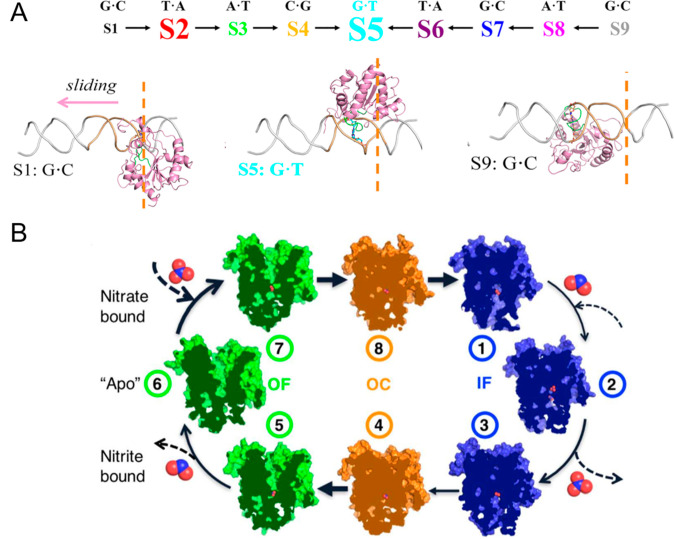
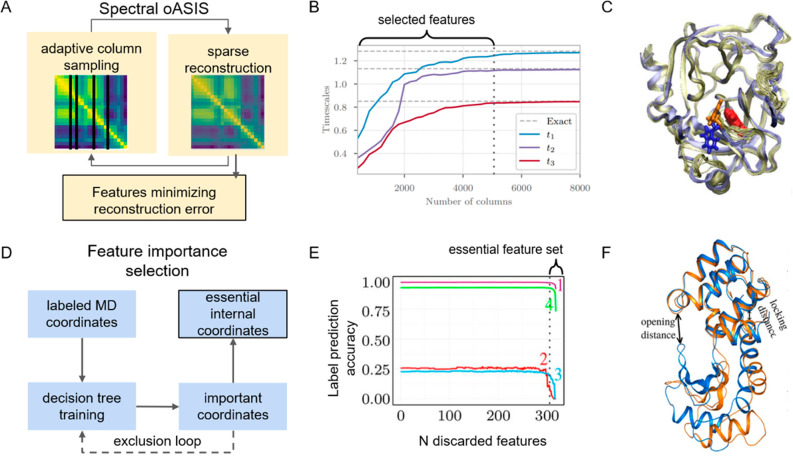
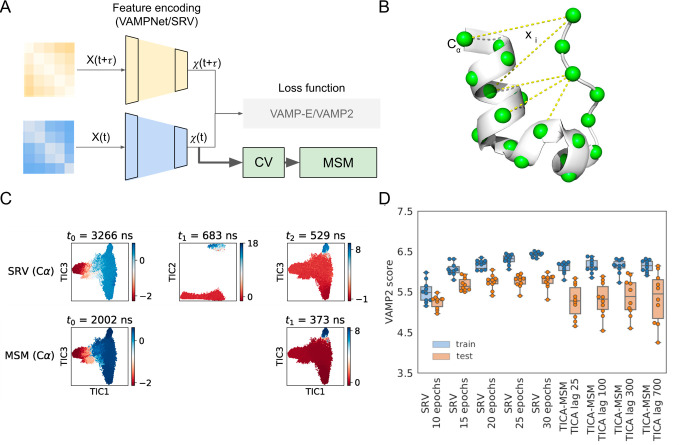
Markov state models (MSMs) based on molecular dynamics (MD) simulations are routinely employed to study protein folding, however, their application to functional conformational changes of biomolecules is still limited. In the past few years, the field of computational chemistry has experienced a surge of advancements stemming from machine learning algorithms, and MSMs have not been left out. Unlike global processes, such as protein folding, the application of MSMs to functional conformational changes is challenging because they mostly consist of localized structural transitions. Therefore, it is critical to properly select a subset of structural features that can describe the slowest dynamics of these functional conformational changes. To address this challenge, we recommend several automatic feature selection methods such as Spectral-OASIS. To identify states in MSMs, the chosen features can be subject to dimensionality reduction methods such as TICA or deep learning based VAMPNets to project MD conformations onto a few collective variables for subsequent clustering. Another challenge for the application of MSMs to the study of functional conformational changes is the ability to comprehend their biophysical mechanisms, as MSMs built for these processes often require a large number of states. We recommend the recently developed quasi-MSMs (qMSMs) to address this issue. Compared to MSMs, qMSMs encode the non-Markovian dynamics via the generalized master equation and can significantly reduce the number of states. As a result, qMSMs can be built with a handful of states to facilitate the interpretation of functional conformational changes. In the wake of machine learning, we believe that the rapid advancement in the MSM methodology will lead to their wider application in studying functional conformational changes of biomolecules.
© 2021 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Elucidating molecular mechanisms of functional conformational changes of proteins via Markov state models.Curr Opin Struct Biol. 2021 Apr;67:69-77. doi: 10.1016/j.sbi.2020.10.005. Epub 2020 Oct 27. Curr Opin Struct Biol. 2021. PMID: 33126140 Review.
-
On the advantages of exploiting memory in Markov state models for biomolecular dynamics.J Chem Phys. 2020 Jul 7;153(1):014105. doi: 10.1063/5.0010787. J Chem Phys. 2020. PMID: 32640825
-
Markov state models provide insights into dynamic modulation of protein function.Acc Chem Res. 2015 Feb 17;48(2):414-22. doi: 10.1021/ar5002999. Epub 2015 Jan 3. Acc Chem Res. 2015. PMID: 25625937 Free PMC article. Review.
-
Tutorial on how to build non-Markovian dynamic models from molecular dynamics simulations for studying protein conformational changes.J Chem Phys. 2024 Mar 28;160(12):121501. doi: 10.1063/5.0189429. J Chem Phys. 2024. PMID: 38516972 Free PMC article.
-
What Markov State Models Can and Cannot Do: Correlation versus Path-Based Observables in Protein-Folding Models.J Chem Theory Comput. 2021 May 11;17(5):3119-3133. doi: 10.1021/acs.jctc.0c01154. Epub 2021 Apr 27. J Chem Theory Comput. 2021. PMID: 33904312 Free PMC article.
Cited by
-
GraphVAMPnets for uncovering slow collective variables of self-assembly dynamics.J Chem Phys. 2023 Sep 7;159(9):094901. doi: 10.1063/5.0158903. J Chem Phys. 2023. PMID: 37655771 Free PMC article.
-
Molecular Mechanisms Underlying the Loop-Closing Dynamics of β-1,4 Galactosyltransferase 1.J Chem Inf Model. 2025 Jan 13;65(1):390-401. doi: 10.1021/acs.jcim.4c02010. Epub 2024 Dec 31. J Chem Inf Model. 2025. PMID: 39737871
-
Extended Quality (eQual): Radial Threshold Clustering Based on n-ary Similarity.J Chem Inf Model. 2025 May 26;65(10):5062-5070. doi: 10.1021/acs.jcim.4c02341. Epub 2025 May 1. J Chem Inf Model. 2025. PMID: 40309753 Free PMC article.
-
Unsupervised and supervised AI on molecular dynamics simulations reveals complex characteristics of HLA-A2-peptide immunogenicity.Brief Bioinform. 2023 Nov 22;25(1):bbad504. doi: 10.1093/bib/bbad504. Brief Bioinform. 2023. PMID: 38233090 Free PMC article.
-
AMUSET-TICA: A Tensor-Based Approach for Identifying Slow Collective Variables in Biomolecular Dynamics.J Chem Theory Comput. 2025 May 13;21(9):4855-4866. doi: 10.1021/acs.jctc.5c00076. Epub 2025 Apr 20. J Chem Theory Comput. 2025. PMID: 40254940
References
-
- Zimmerman M. I.; Porter J. R.; Ward M. D.; Singh S.; Vithani N.; Meller A.; Mallimadugula U. L.; Kuhn C. E.; Borowsky J. H.; Wiewiora R. P.; Hurley M. F. D.; Harbison A. M.; Fogarty C. A.; Coffland J. E.; Fadda E.; Voelz V. A.; Chodera J. D.; Bowman G. R. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. 10.1038/s41557-021-00707-0. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous