

CDKL5 kinase controls transcription-coupled responses to DNA damage
- PMID: 34605059
- PMCID: PMC8634139
- DOI: 10.15252/embj.2021108271
CDKL5 kinase controls transcription-coupled responses to DNA damage
Abstract
Mutations in the gene encoding the CDKL5 kinase are among the most common genetic causes of childhood epilepsy and can also give rise to the severe neurodevelopmental condition CDD (CDKL5 deficiency disorder). Despite its importance for human health, the phosphorylation targets and cellular roles of CDKL5 are poorly understood, especially in the cell nucleus. Here, we report that CDKL5 is recruited to sites of DNA damage in actively transcribed regions of the nucleus. A quantitative phosphoproteomic screen for nuclear CDKL5 substrates reveals a network of transcriptional regulators including Elongin A (ELOA), phosphorylated on a specific CDKL5 consensus motif. Recruitment of CDKL5 and ELOA to damaged DNA, and subsequent phosphorylation of ELOA, requires both active transcription and the synthesis of poly(ADP-ribose) (PAR), to which CDKL5 can bind. Critically, CDKL5 kinase activity is essential for the transcriptional silencing of genes induced by DNA double-strand breaks. Thus, CDKL5 is a DNA damage-sensing, PAR-controlled transcriptional modulator, a finding with implications for understanding the molecular basis of CDKL5-related diseases.
Keywords: CDKL5 disorder; DNA damage response; kinase; poly(ADP-ribose); transcriptional regulation.
© 2021 The Authors. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
The authors declare no conflict of interests.
Figures
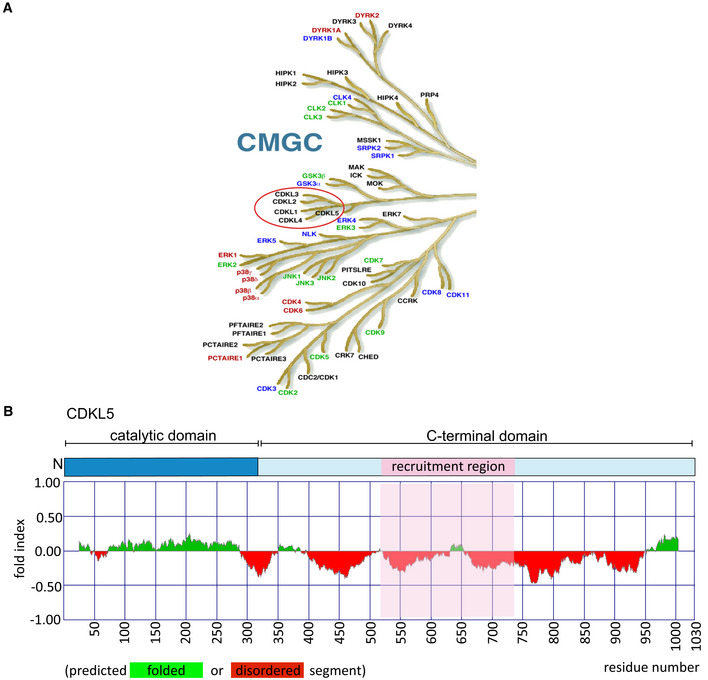
The CMGC branch of the human kinome. The figure is taken from the dendrogram made by Manning and colleagues (Manning et al, 2002). The CDKL family of kinases that includes CDKL5 is encircled in red.
(Top) Schematic diagram of CDKL5 protein. (Bottom) Bioinformatics analysis for CDKL5 folding using Fold index software (Prilusky et al, 2005). The plot shows the disorder prediction for CDKL5 protein sequence. Ordered regions are indicated in green above 0, while disordered regions are indicated in red below 0. Amino acids suggested as being folded or unfolded are depicted at the bottom of the plot. The region that mediates recruitment of CDKL5 to DNA damage sites is marked in pink.
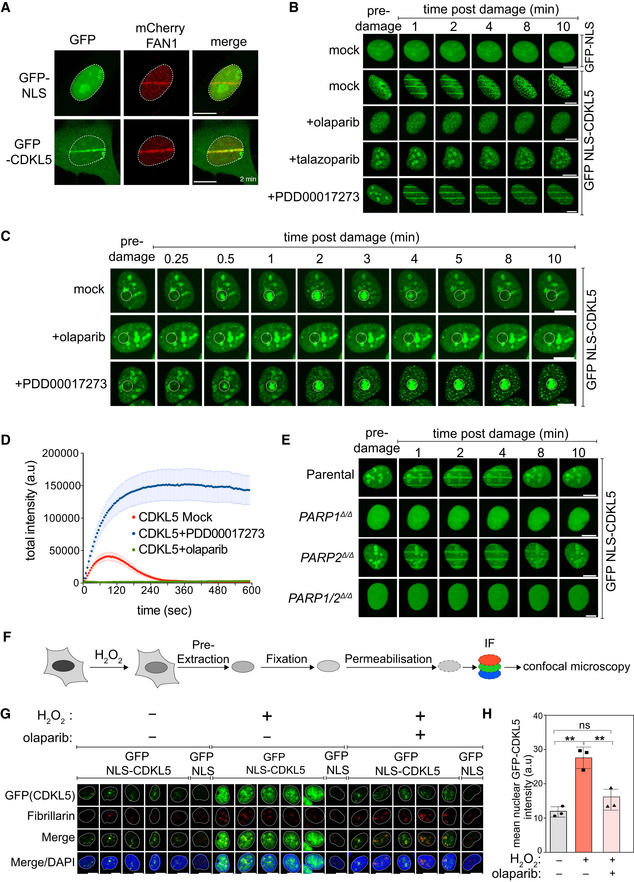
BrdU‐sensitized U‐2‐OS Flp‐In T‐REx cells stably expressing mCherry‐FAN1 and GFP‐NLS or GFP–CDKL5 (no NLS) were line‐micro‐irradiated (355 nm) and imaged after 2 min. Scale bar is 10 μm.
BrdU‐sensitized U‐2‐OS cells stably expressing GFP‐NLS‐CDKL5 were pre‐incubated with DMSO (mock), olaparib (5 μM), talazoparib (50 nM) or PDD00017273 (0.3 μM) for 1 h prior to micro‐irradiation and live imaged at the indicated times post–irradiation. One of three independent experiments is shown. Scale bar is 10 μm.
Same as (B) except that cells stably expressing GFP‐NLS‐CDKL5 were pre‐incubated with DMSO (mock), olaparib (5 μM) or PDD00017273 (0.3 μM) for 1 h prior to spot micro‐irradiation (405 nm). Individual cells from one of two independent biological replicates are shown. Scale bar is 10 μm.
Quantitation of spot intensities. Data represent the mean ± SEM of two independent experiments; > 50 micro‐irradiated cells per point.
BrdU‐sensitized parental or PARP1 Δ/Δ, PARP2 Δ/Δ, PARP1/2 Δ/Δ U–2–OS cells transiently expressing GFP‐NLS‐CDKL5 were subjected to 355 nm line micro‐irradiation followed by time‐lapse imaging. One of two independent experiments is shown. Scale bar is 10 μm.
Diagram of the workflow for the chromatin retention experiments.
Cells subjected to the workflow in (F) were detergent–extracted and fixed before staining with anti‐GFP or fibrillarin (nucleoli). Scale bar is 10 μm.
Quantification of the detergent‐insoluble GFP‐NLS‐CDKL5 signal (minus nucleolar signal). The mean ± SD from three biological experiments is shown. Statistical significance was assessed by one‐way ANOVA test. Asterisks ** indicate P‐value of < 0.01; ns—not significant.
- A
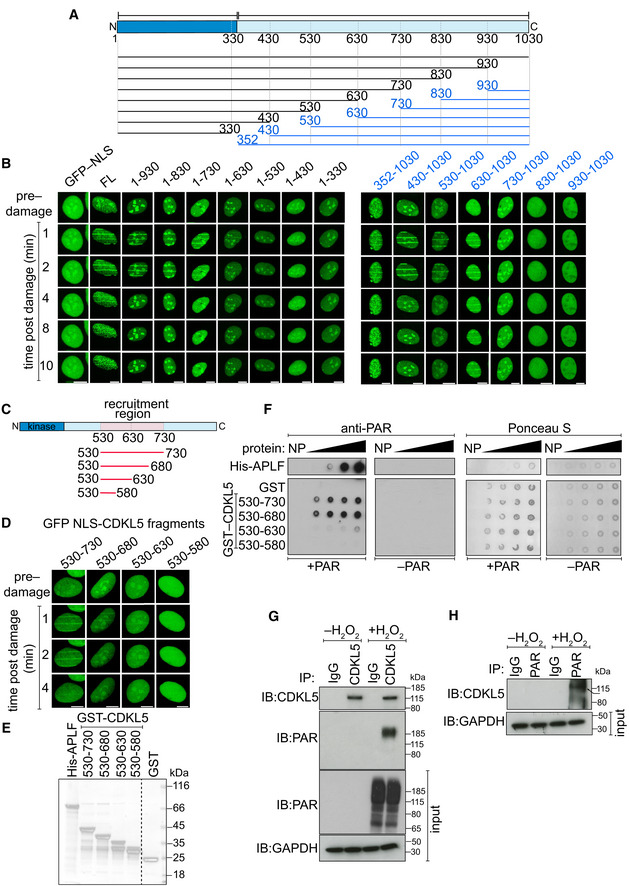
Schematic diagram of CDKL5 deletion mutants, deleting from the N‐terminal (blue) or C‐terminal (black) ends. All proteins were expressed with an N‐terminal NLS and GFP tag.
- B
BrdU‐sensitized U‐2‐OS (Flp‐In T‐REx) cells stably expressing GFP‐NLS, the GFP‐NLS‐CDKL5 deletion mutants shown in (A) or full length (FL) GFP‐NLS‐CDKL5 was subjected to line micro‐irradiation (355 nm) and time‐lapse imaging. Three independent experiments were performed, and one representative experiment is shown. Scale bar is 10 μm.
- C
Schematic for fragments corresponding to the PAR‐dependent recruitment region in CDKL5 as identified in (B).
- D
Same as in (B) except that the GFP‐NLS‐tagged CDKL5 fragments indicated were examined. Scale bar is 10 μm.
- E
Coomassie gel showing recombinant fragments of human CDKL5 fused to GST purified from bacterial lysates. GST and APLF were also purified as controls.
- F
Recombinant fragments of CDKL5 fused to GST (1.2, 2.5, 5, 10 µg), or GST, were dot‐blotted on nitrocellulose membrane and then incubated with synthetic PAR. PAR binding was detected by far Western blotting. APLF was used as positive control. One of three independent experiments is shown.
- G, H
U‐2‐OS (Flp‐In T‐Rex) cells stably expressing CDKL5 were either mock‐treated or treated with 500 µM H2O2 for 30 min. Extracts were subjected to immunoprecipitation with antibodies against CDKL5 (G) or PAR (H) (or non‐specific IgG as control). Precipitates, and input lysates, were analysed by Western blotting using the indicated antibodies. One of two independent experiments is shown.
- A
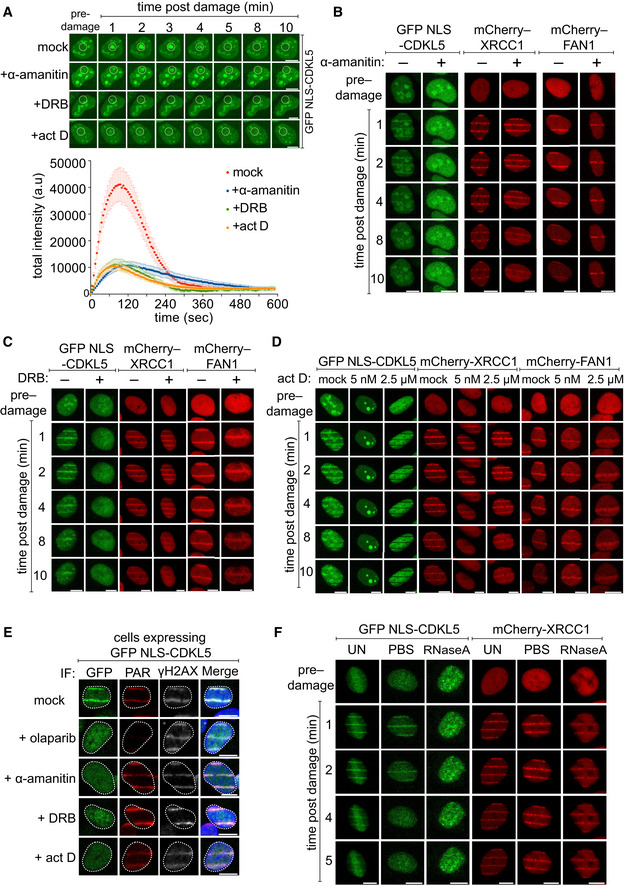
(Top) BrdU‐sensitized U‐2‐OS (Flp‐In T‐REx) cells stably expressing GFP‐NLS‐CDKL5 were treated with indicated transcription inhibitors before subjecting to spot micro‐irradiation (405 nm). (Bottom) Quantitation of spot intensities. Data represent the mean ± SEM of two independent experiments; > 50 micro‐irradiated cells per point. The “mock” trace shown is identical to the “mock” trace shown in Fig 1D, as the data come from the same set of biological replicates. Scale bar is 10 μm.
- B–D
BrdU‐sensitized U‐2‐OS (Flp‐In T‐Rex) cells stably expressing GFP‐NLS‐CDKL5, mCherry‐XRCC1 or mCherry‐FAN1 were pre‐incubated with indicated transcription inhibitors prior to line micro‐irradiation (355 nm) and time‐lapse imaging. One of three independent experiments is shown. Scale bar is 10 μm.
- E
Same as (B–D) except that BrdU‐sensitized cells stably expressing GFP‐NLS‐CDKL5 were also pre‐incubated with olaparib as control. Cells were subjected to line micro‐irradiation, fixed and then subjected to indirect immunofluorescence using antibodies against GFP, PAR and γH2AX. Scale bar is 10 μm.
- F
Stable cell lines were permeabilized and incubated with RNase A or PBS before irradiation and imaging. Scale bar is 10 μm.
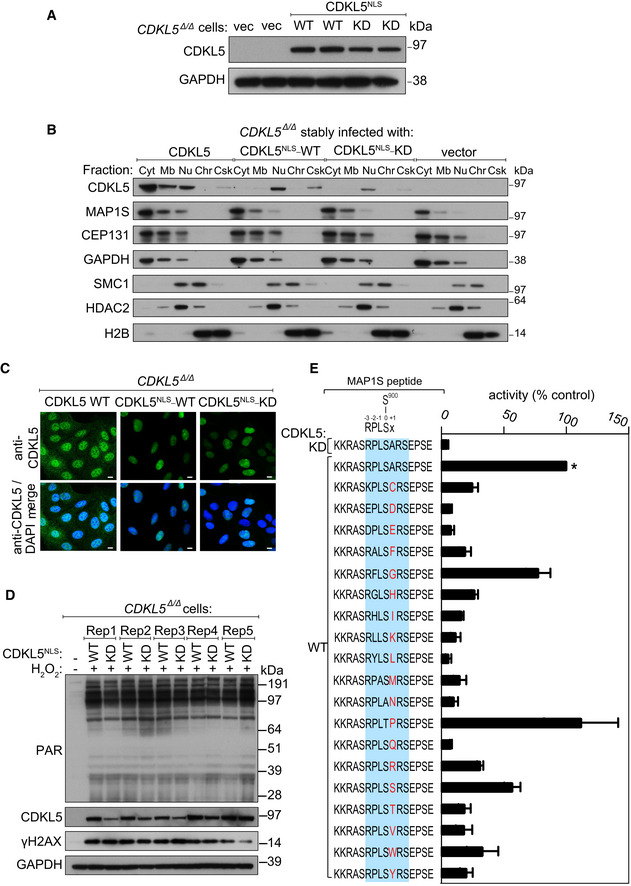
Extracts of CDKL5‐disrupted U‐2‐OS (Flp‐In T‐REx) cells (CDKL5 Δ/Δ) stably expressing CDKL5NLS WT or a K42R kinase‐dead mutant (CDKL5NLS–KD) or empty vector were subjected to Western blotting with the antibodies indicated. Two different dishes of cells are shown per condition.
Subcellular fractionation of lysates from CDKL5 Δ/Δ cells stably expressing CDKL5, CDKL5NLS WT or CDKL5NLS KD or empty vector. Lysates were fractionated to isolate proteins found in the following subcellular compartments: cytoplasmic (Cyt), membrane (Mb), nuclear (Nuc), chromatin (Ch) or cytoskeleton (Csk). Fractionated samples were resolved by SDS–PAGE and probed with antibodies shown.
CDKL5 Δ/Δ cells stably expressing CDKL5, CDKL5NLS WT or CDKL5NLS KD were subjected to indirect immunofluorescence analysis with anti‐CDKL5 antibodies. Scale bar is 10 μm.
CDKL5 Δ/Δ cells stably expressing CDKL5NLS WT or CDKL5NLS KD (or empty vector) were treated with 500 µM H2O2 for 15 min. Samples were resolved by SDS–PAGE and probed with indicated antibodies or stained with Ponceau S to show equal loading. Rep=biological replicate.
Peptide kinase assays to investigate CDKL5 sequence specificity. Anti‐FLAG precipitates from HEK293 cells transiently expressing FLAG‐tagged CDKL5 (wild‐type “WT” or a K42R kinase‐dead “KD” mutant) were incubated with synthetic peptides corresponding to sequence around the previously reported CDKL5 phosphorylation site in MAP1S (Ser900) designed specifically to investigate the effect of amino acid substitutions A901 on the phosphorylation of MAP1S Ser900. Assays were done in the presence of [γ‐32P]‐labelled ATP‐Mg2+, and peptide phosphorylation was measured by Cerenkov counting. Phosphorylation of the control wild‐type MAP1S peptide is taken as 100% (*). The data are represented as mean ± SEM from three independent experiments. The RPXSA motif is shaded in blue, and amino acid substitutions compared with the wild‐type MAP1S Ser900 peptide are shown in red.
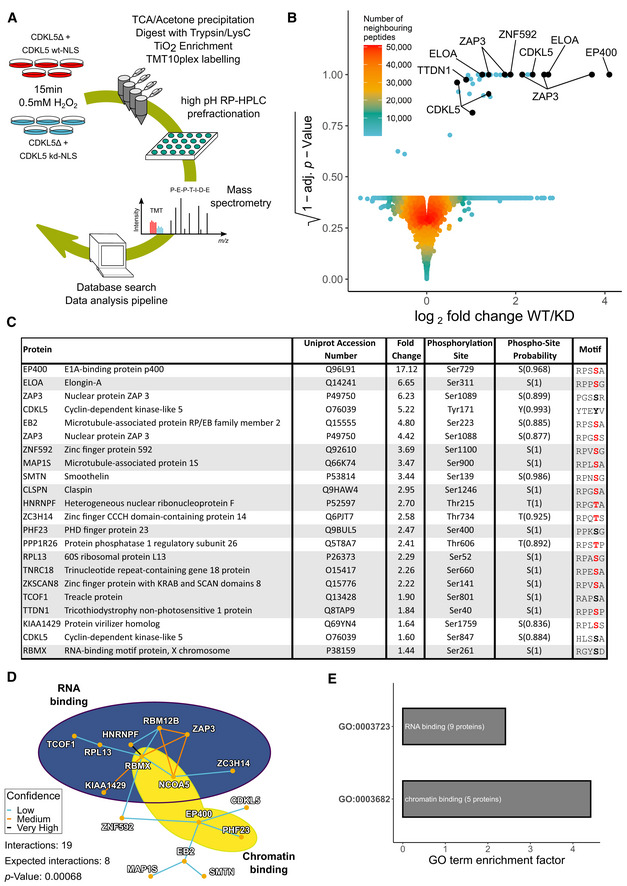
Quantitative phosphoproteomic workflow.
Volcano plot of the mass spectrometric data from the experiment in (A) (see Table EV1).
List of proteins containing phosphorylation sites more abundant in cells expressing CDKL5NLS WT vs KD, with phosphorylation sites having PTM score probabilities > 75% (peptides with a 100% PTM score probability are shaded in grey). Phosphorylation sites with a R–P–X–[S/T]–[A/G/P/S] motif are highlighted in red.
Protein–protein interaction network of putative CDKL5 substrates from Table EV1. Confidence levels are based on the STRING database v11.0 combined score with following bins: 150–400: low confidence (blue); 400–700: medium confidence (gold); 700–900: high confidence (not encountered in this dataset); and > 900: very high confidence (black). P‐value was calculated as 0.00068.
Analysis of GO terms. Significance cut‐off was set as α = 0.01 with at least three proteins identified in the respective group. GO term enrichment factor expresses the relative over‐representation of the GO term within the group of proteins containing a phosphorylation site that is more abundant in WT compared with KD compared with the group of all proteins.
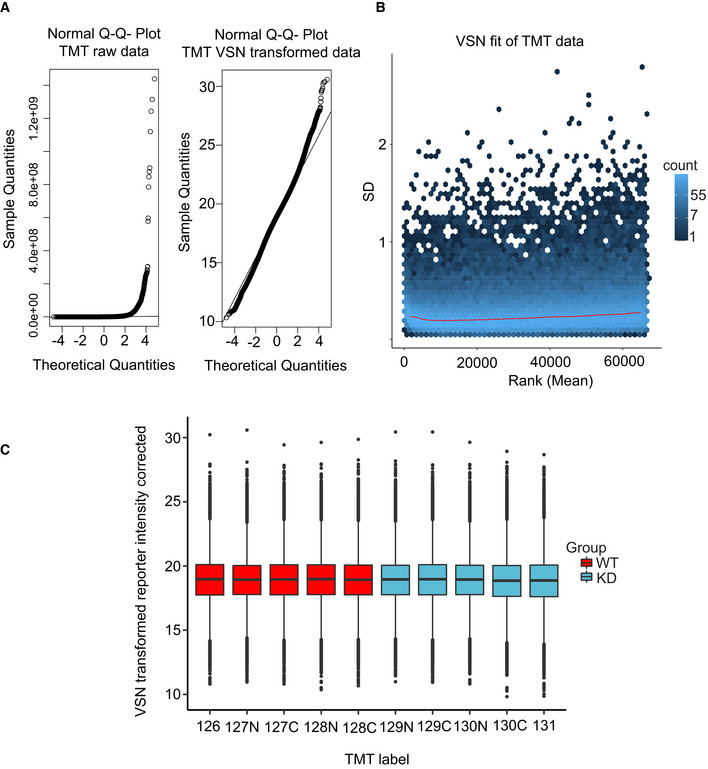
Left: Normal Q–Q plot of the raw TMT intensity data with large deviations from a normal distribution as seen from datapoints not following the indicated line in the plot. Right: Q–Q plot of the TMT intensity data after VSN transformation. Only minor deviations from the line indicates the transformed data follow a normal distribution to a satisfactory degree. The hypervariable datapoints in the upper quantiles are controlled by the application of the robust implementation of the empirical Bayes algorithm used by limma (Phipson et al, 2016) and implemented in the analysis scripts.
Standard deviation plotted against the intensity rank of the VSN‐transformed TMT data. Red line indicates the mean standard deviation. Line is approximately horizontal, indicating that the variance is not overly dependent on intensity rank and suggests a successful VSN transform.
Boxplot of intensity distribution in each TMT channel. No obvious discrepancy between the median values of the individual channels indicates a successful calibration by VSN and no introduction of an obvious intensity bias for any experimental group. The central band of the boxplot indicates the median value, while the hinges represent the first and third quartile (bottom and top of boxplot, respectively). The whiskers extend to the largest/smallest (upper or lower whisker, respectively) datapoint not further than 1.5 times the interquartile range from their respective hinge. The experiment was conducted using five biological replicates of CDKL5NLS WT (WT, red) and CDKL5NLS KD (KD, blue) where each TMT channel represents a single biological replicate from the respective group.
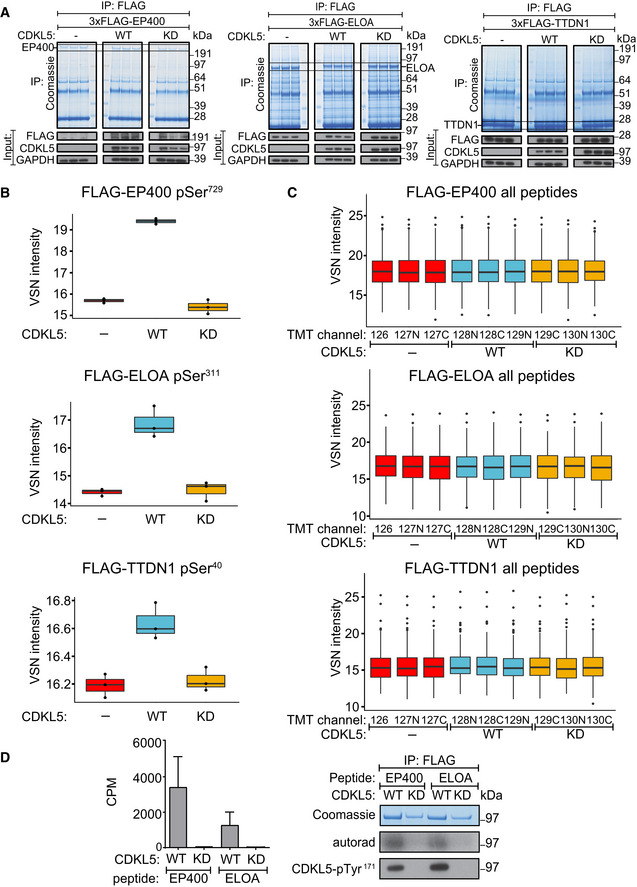
HEK293 cells were co‐transfected with CDKL5NLS (wild‐type “WT” or kinase‐dead “KD” K42R mutant) and either FLAG‐EP400 (left), FLAG‐ELOA (middle) or FLAG‐TTDN1 (right). 24 h later, cells were incubated with H2O2 (500 µM) for 15 min before being harvested and lysed. Protein extracts were subjected to immunoprecipitation with anti‐FLAG‐agarose beads. Precipitates were subjected to SDS–PAGE and blotting with antibodies shown (bottom panels) or staining with Coomassie Brilliant Blue (top panels). The bands corresponding to the FLAG‐tagged proteins were excised from the gels in A. and processed for mass spectrometric detection of relevant phosphopeptides. Three independent co‐transfection experiments were done for every condition.
Boxplots showing VSN‐normalized intensity of phosphopeptides corresponding to EP400‐pSer729, ELOA‐pSer311 and TTDN1‐pSer40 from the experiment in (A). The central band of the boxplot indicates the median value, while the hinges represent the first and third quartile (bottom and top of boxplot, respectively). The whiskers extend to the largest/smallest (upper or lower whisker, respectively) datapoint not further than 1.5 times the interquartile range from their respective hinge. In all cases, the data were derived from 3 biological replicates.
Boxplots of the VSN‐adjusted TMT reporter ion intensities for all peptides for each TMT label in the case of FLAG‐EP400, FLAG‐ELOA and FLAG‐TTDN1 from the experiment in (A). The central band of the boxplot indicates the median value, while the hinges represent the first and third quartile (bottom and top of boxplot, respectively). The whiskers extend to the largest/smallest (upper or lower whisker, respectively) datapoint not further than 1.5 times the interquartile range from their respective hinge. Datapoints were further removed, and then, the whiskers are plotted individually. The experiment was conducted using three biological replicates within each respective group, and each TMT channel represents a single biological replicate.
Left: Anti‐FLAG precipitates from HEK293 cells transiently expressing FLAG‐tagged CDKL5 (wild‐type “WT” or a K42R kinase‐dead “KD” mutant) were incubated with the synthetic peptides indicated, in the presence of [γ‐32P]‐labelled ATP‐Mg2+, and peptide phosphorylation was measured by the Cerenkov counting. Data are represented as mean ± SEM from three independent experiments. Right: Same but anti‐FLAG precipitates were subjected to SDS–PAGE and autoradiography to detect CDKL5 autophosphorylation, or Western blotting with CDKL5‐pTyr171 antibody specific for the CDKL5‐Tyr171 autophosphorylation site (Munoz et al, 2018).
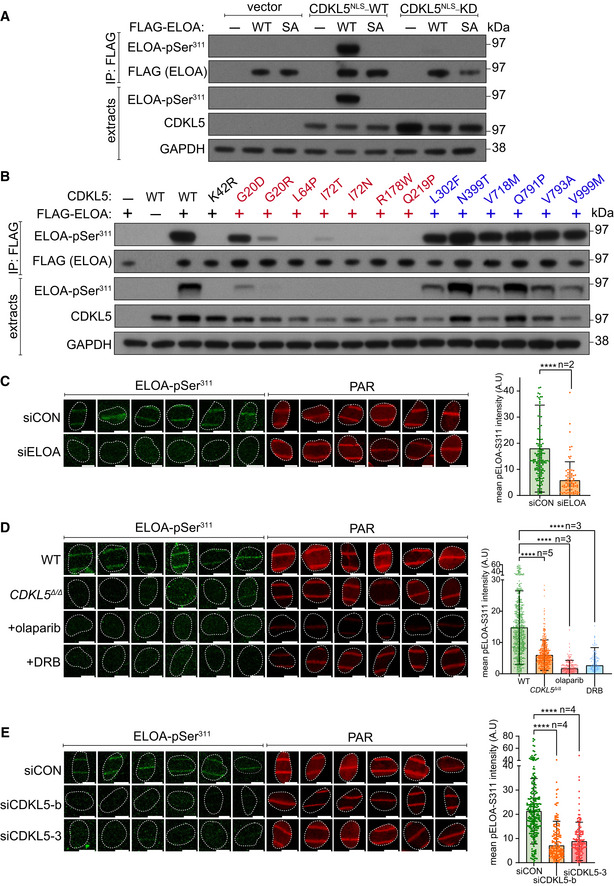
- A
HEK293 cells were co‐transfected with CDKL5 (wild‐type “WT” or kinase‐dead “KD” K42R mutant) fused to an NLS, and FLAG‐ELOA (wild‐type “WT” or a S311A mutant “SA”). Anti‐FLAG precipitates or cell extracts were probed with the antibodies indicated. One of three independent experiments is shown.
- B
Same as (A) showing a range of pathogenic (red) and benign (blue) CDKL5 variants.
- C–E
Wild‐type (WT), CDKL5‐disrupted (CDKL5Δ / Δ) or siRNA‐transfected cells were subjected to indirect immunofluorescence analysis with the indicated antibodies at laser tracks. Quantification of ELOA‐pSer311 signal at the laser tracks is shown. Data represent mean ± SD of total pELOA Ser311 intensities in different biological replicates as indicated (n). For simplicity, only intensities greater than zero are shown. Statistical significance was assessed by one‐way ANOVA test or the unpaired t‐test with Welch's correction. Asterisks **** indicate P‐values of < 0.0001. Scale bar is 10 μm.
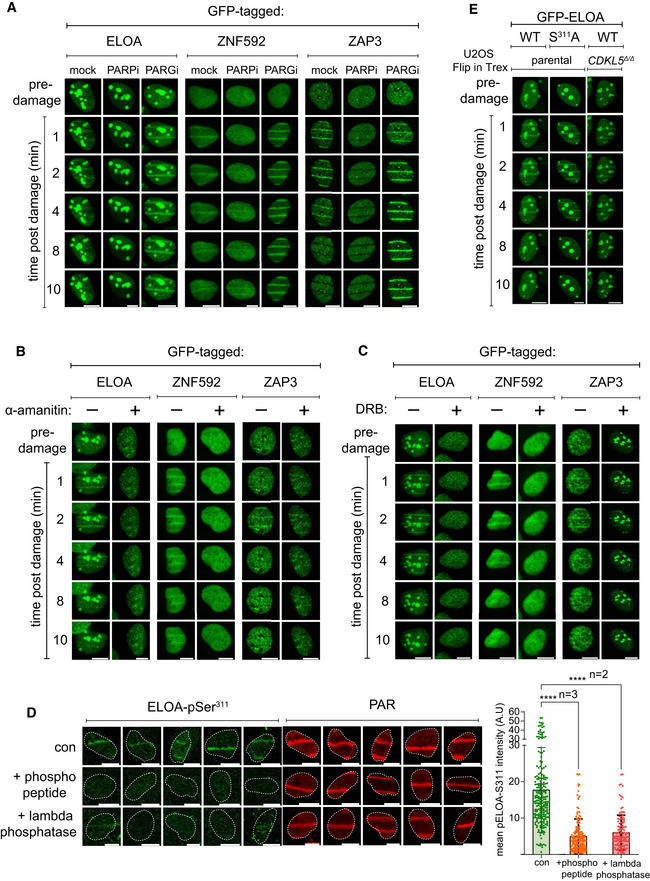
- A–C
BrdU‐sensitized U‐2‐OS (Flp‐In T‐REx) cells stably expressing GFP‐tagged forms of the proteins indicated were pre‐incubated with (A) olaparib (PARPi; 5 µM) or PD00017273 (PARGi; 0.3 µM, 1 h), (B) α‐amanitin (20 µg/ml, 8 h) or (C) DRB (100 µM, 2 h) prior to line micro‐irradiation (355 nm) and time‐lapse imaging. One of three independent experiments is shown. Scale bar is 10 μm.
- D
BrdU‐sensitized U‐2‐OS (Flp‐In T‐REx) cells were subjected to nuclear line micro‐irradiation (355 nm). Cells were fixed and then mock‐treated (con) or treated with lambda‐phosphatase prior to incubation with the primary antibodies indicated. Alternatively, ELOA‐pSer311 phosphopeptide was included during incubation with the primary antibodies before indirect immunofluorescence analysis. Quantification of ELOA‐pSer311 signal at the laser tracks is shown. Data represent mean ± SD of total pELOA Ser311 intensities in different biological replicates as indicated (n). For simplicity, only intensities greater than zero are shown. Statistical significance was assessed by one‐way ANOVA test. Asterisks **** indicate P‐values of < 0.0001. Scale bar is 10 μm.
- E
BrdU‐sensitized U‐2‐OS cells (Flp‐In T‐Rex; CDKL5–disrupted (CDKL5Δ / Δ) or parental cells) stably expressing GFP‐tagged ELOA wild‐type (WT) or S311A mutant were line‐micro‐irradiated and imaged after at the time points indicated.
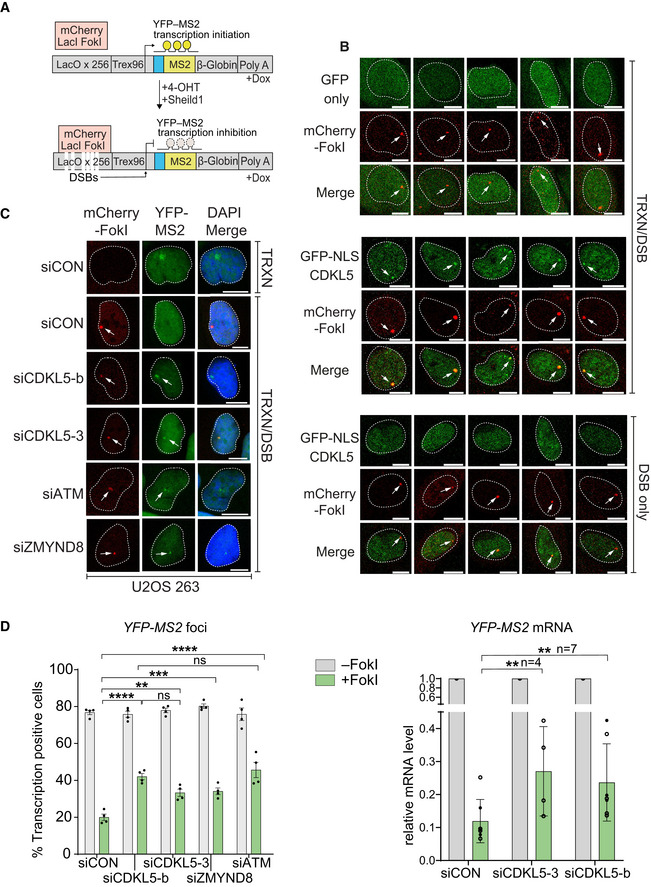
Cartoon of reporter construct (Tang et al, 2013) in which induction of the mCherry‐tagged FokI endonuclease (with 4‐OHT) results in double‐strand break (DSB) in a region upstream of a doxycycline‐inducible reporter gene (YFP‐MS2). Ongoing transcription of the reporter gene can be visualized by the presence of a YFP‐MS2 fusion protein that binds stem–loop structures in the nascent transcript.
CDKL5 is recruited to FokI‐induced DSBs. GFP alone (top panel) or GFP‐NLS‐CDKL5 (middle and bottom panels) was stably expressed in U‐2‐OS 265 DSB reporter cells. Cells were mock‐treated or treated with 1 µg/ml doxycycline for 3 h to induce transcription of the reporter gene. An hour before DSB induction, cells were treated with 0.3 µM PARG and 10 µM ATM inhibitor. Site‐specific DSBs were induced by treating the cells with 4‐OHT and Sheild1 ligand. Cells were live‐imaged at 37°C, between 15 and 25 min following DSB induction. Representative image showing the recruitment of GFP‐NLS‐CDKL5 to FokI‐induced DSBs upstream of transcriptionally active (middle) but not the inactive (bottom) MS2 gene. GFP alone is used a control (top). White arrowheads mark the location of the mCherry‐FokI upstream of the MS2 reporter cassette. Images are representative of multiple technical replicates of three independent experiments. Scale bar is 10 μm.
Representative image for U–2–OS 263 IFII cells harbouring the reporter construct and transfected with the siRNAs indicated. After addition of doxycycline, transcription was monitored in cells ± induction of FokI by quantification of YFP(–MS2) foci. Arrows indicate sites of FokI‐mediated DSB (mCherry) and YFP‐MS2 transcript. “TRXN”: doxycycline added; “TRXN/DSB”: doxycycline added with 4‐OHT. Scale bar is 10 μm.
(Left) Quantification of transcription in U‐2‐OS 263 IFII reporter cells from experiment in B. > 150 cells were analysed per condition per experiment. The mean ± SD from four independent experiments is shown. (Right) Quantitative RT–PCR analysis of YFP‐MS2 mRNA in U‐2‐OS 263 reporter cells. Data represent mean ± SD in different biological replicates as indicated (n). Statistical significance for all the data was assessed by two‐way ANOVA test, **P < 0.01, ***P < 0.001 and ****P < 0.0001; ns—not significant.
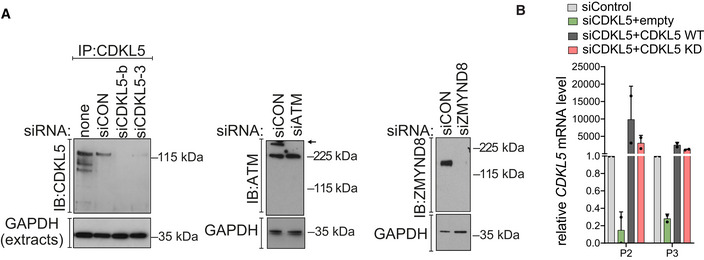
CDKL5, ATM and ZMYND8 were depleted in U‐2‐OS 263 IFII reporter cells using indicated siRNA. siCON—non‐targeting control.
qRT–PCR measurements in U2OS I‐PpoI cells to validate the silencing efficiency of siCDKL5 and the subsequent rescue efficiency of ectopic expression of siRNA‐resistant forms of CDKL5 wild‐type (WT) and K42R kinase‐dead mutant (KD). P2 and P3 primer pairs were used to analyse the changes in mRNA levels. The mean ± SD from two qPCR replicates of two independent experiments is shown.
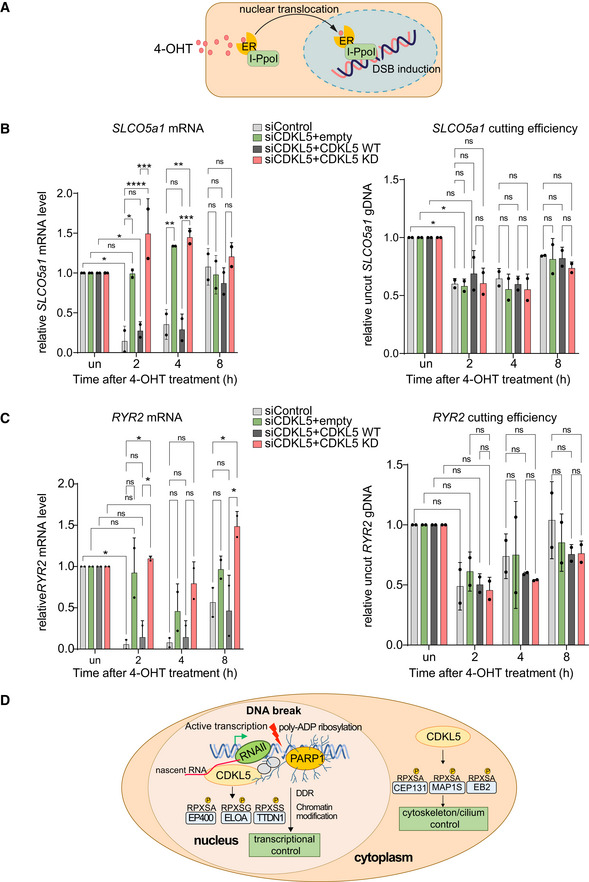
- A
Schematic diagram of the I‐PpoI system for inducing DNA breaks in the nuclear human genome. Addition of 4‐OHT to U‐2‐OS‐pEP15 cells stably expressing the I‐PpoI endonuclease fused to the estrogen receptor (ER) induces nuclear translocation of the fusion protein and cleavage cleavage of FokI recognition sites in nuclear DNA resulting, leading to DSB induction.
- B, C
Quantitative PCR with reverse transcription (qRT–PCR) analysis of SLCO5a1 (B) and RYR2 expression levels (C) (left panels) U‐2‐OS HA‐ER‐I‐PpoI cells depleted of CDKL5 transiently transfected with FLAG‐tagged CDKL5 wild‐type (WT) or a K42R‐mutated kinase‐dead (KD) mutant, or empty vector, at the times indicated after inducing I‐PpoI. The mean ± SD from two qPCR replicates of two independent experiments is shown. Statistical significance for all the data was assessed by two‐way ANOVA test *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; ns—not significant. I‐PpoI‐mediated cutting efficiency in the relevant gene is shown in the right‐hand panel (see Materials and Methods).
- D
Schematic diagram depicting CDKL5 functions in nucleus and cytosol.
References
-
- Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC (2008) Poly(ADP‐ribose)‐binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451: 81–85 - PubMed
-
- Aleksandrov R, Dotchev A, Poser I, Krastev D, Georgiev G, Panova G, Babukov Y, Danovski G, Dyankova T, Hubatsch L et al (2018) Protein dynamics in complex DNA lesions. Mol Cell 69: 1046–1061.e5 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
