

Assessing deep learning methods in cis-regulatory motif finding based on genomic sequencing data
- PMID: 34607350
- PMCID: PMC8769700
- DOI: 10.1093/bib/bbab374
Assessing deep learning methods in cis-regulatory motif finding based on genomic sequencing data
Abstract
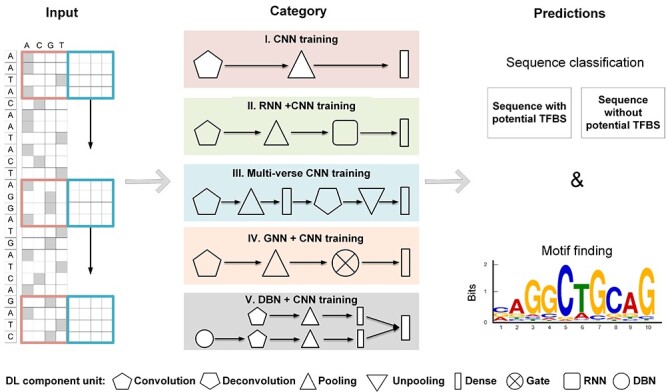
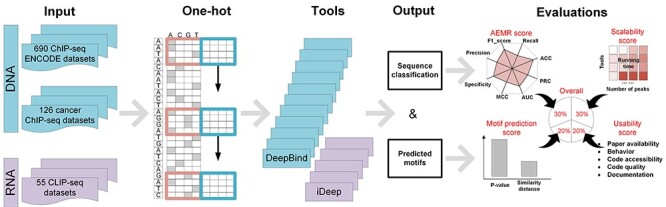
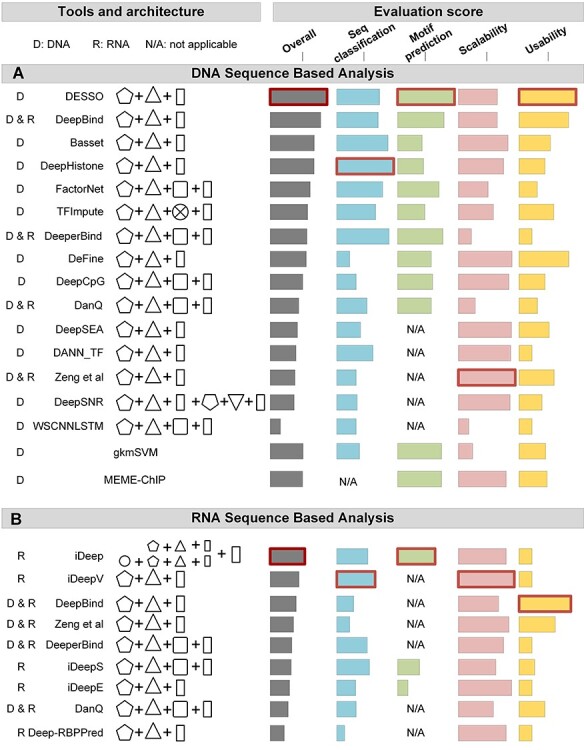
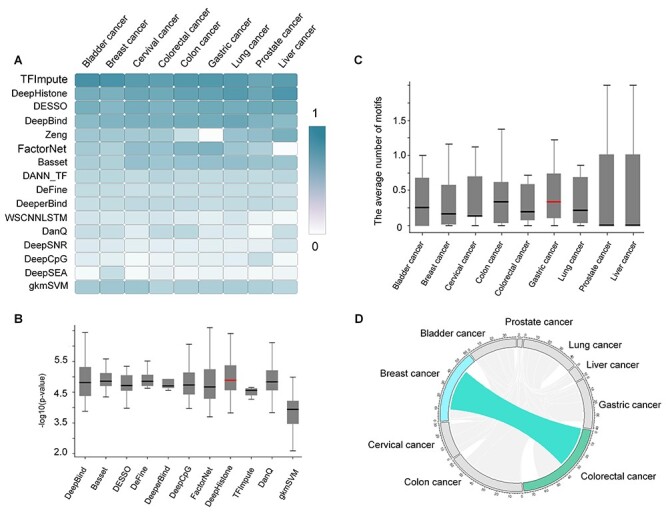
Identifying cis-regulatory motifs from genomic sequencing data (e.g. ChIP-seq and CLIP-seq) is crucial in identifying transcription factor (TF) binding sites and inferring gene regulatory mechanisms for any organism. Since 2015, deep learning (DL) methods have been widely applied to identify TF binding sites and predict motif patterns, with the strengths of offering a scalable, flexible and unified computational approach for highly accurate predictions. As far as we know, 20 DL methods have been developed. However, without a clear and systematic assessment, users will struggle to choose the most appropriate tool for their specific studies. In this manuscript, we evaluated 20 DL methods for cis-regulatory motif prediction using 690 ENCODE ChIP-seq, 126 cancer ChIP-seq and 55 RNA CLIP-seq data. Four metrics were investigated, including the accuracy of motif finding, the performance of DNA/RNA sequence classification, algorithm scalability and tool usability. The assessment results demonstrated the high complementarity of the existing DL methods. It was determined that the most suitable model should primarily depend on the data size and type and the method's outputs.
Keywords: CLIP-seq; ChIP-seq; TF binding sites identification; deep learning method assessment; motif prediction.
© The Author(s) 2021. Published by Oxford University Press.
Figures
Similar articles
-
An algorithmic perspective of de novo cis-regulatory motif finding based on ChIP-seq data.Brief Bioinform. 2018 Sep 28;19(5):1069-1081. doi: 10.1093/bib/bbx026. Brief Bioinform. 2018. PMID: 28334268 Review.
-
CacPred: a cascaded convolutional neural network for TF-DNA binding prediction.BMC Genomics. 2025 Mar 18;26(Suppl 2):264. doi: 10.1186/s12864-025-11399-y. BMC Genomics. 2025. PMID: 40102719 Free PMC article.
-
Improving analysis of transcription factor binding sites within ChIP-Seq data based on topological motif enrichment.BMC Genomics. 2014 Jun 13;15(1):472. doi: 10.1186/1471-2164-15-472. BMC Genomics. 2014. PMID: 24927817 Free PMC article.
-
De novo prediction of cis-regulatory elements and modules through integrative analysis of a large number of ChIP datasets.BMC Genomics. 2014 Dec 2;15:1047. doi: 10.1186/1471-2164-15-1047. BMC Genomics. 2014. PMID: 25442502 Free PMC article.
-
Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond.Cell Cycle. 2014;13(18):2847-52. doi: 10.4161/15384101.2014.949201. Cell Cycle. 2014. PMID: 25486472 Free PMC article. Review.
Cited by
-
Predicting miRNA-disease associations based on multi-view information fusion.Front Genet. 2022 Sep 27;13:979815. doi: 10.3389/fgene.2022.979815. eCollection 2022. Front Genet. 2022. PMID: 36238163 Free PMC article.
-
Identifying transcription factors with cell-type specific DNA binding signatures.BMC Genomics. 2024 Oct 14;25(1):957. doi: 10.1186/s12864-024-10859-1. BMC Genomics. 2024. PMID: 39402535 Free PMC article.
-
Uncovering the Relationship between Tissue-Specific TF-DNA Binding and Chromatin Features through a Transformer-Based Model.Genes (Basel). 2022 Oct 26;13(11):1952. doi: 10.3390/genes13111952. Genes (Basel). 2022. PMID: 36360189 Free PMC article.
-
MMGAT: a graph attention network framework for ATAC-seq motifs finding.BMC Bioinformatics. 2024 Apr 20;25(1):158. doi: 10.1186/s12859-024-05774-x. BMC Bioinformatics. 2024. PMID: 38643066 Free PMC article.
-
MMGraph: a multiple motif predictor based on graph neural network and coexisting probability for ATAC-seq data.Bioinformatics. 2022 Sep 30;38(19):4636-4638. doi: 10.1093/bioinformatics/btac572. Bioinformatics. 2022. PMID: 35997564 Free PMC article.
References
-
- D'haeseleer P. What are DNA sequence motifs? Nat Biotechnol 2006;24:423–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
