

Gitelman-Like Syndrome Caused by Pathogenic Variants in mtDNA
- PMID: 34607911
- PMCID: PMC8819995
- DOI: 10.1681/ASN.2021050596
Gitelman-Like Syndrome Caused by Pathogenic Variants in mtDNA
Abstract
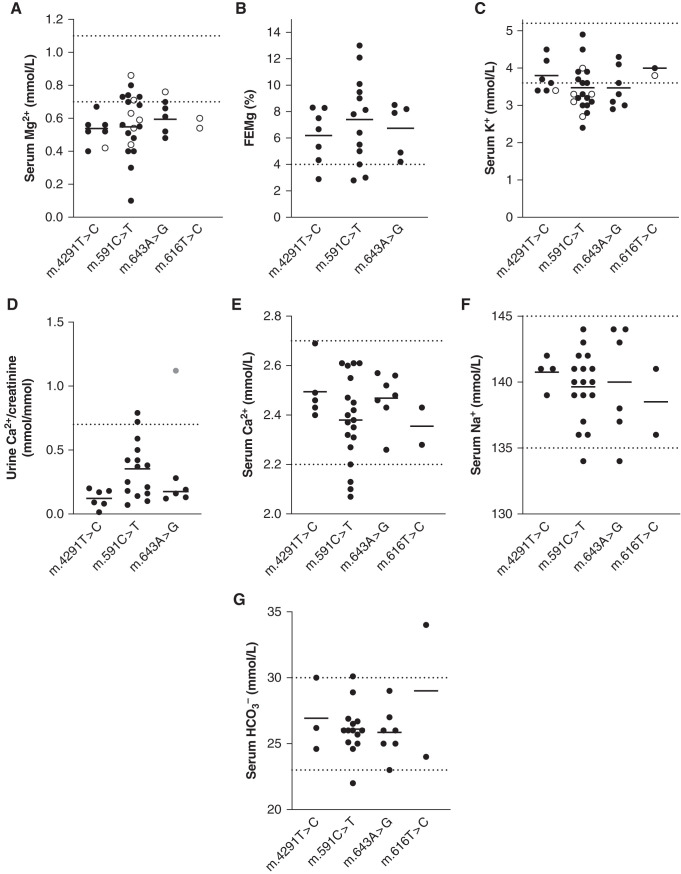
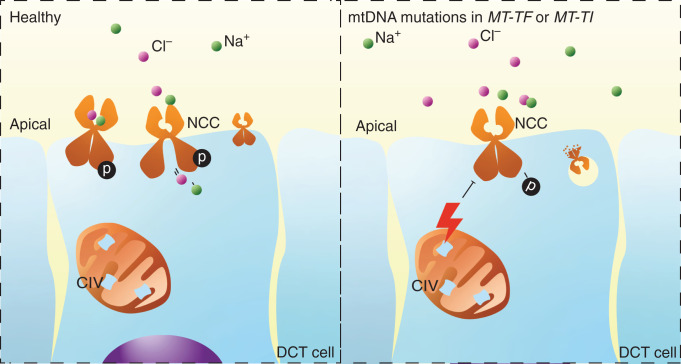
Background: Gitelman syndrome is the most frequent hereditary salt-losing tubulopathy characterized by hypokalemic alkalosis and hypomagnesemia. Gitelman syndrome is caused by biallelic pathogenic variants in SLC12A3, encoding the Na+-Cl- cotransporter (NCC) expressed in the distal convoluted tubule. Pathogenic variants of CLCNKB, HNF1B, FXYD2, or KCNJ10 may result in the same renal phenotype of Gitelman syndrome, as they can lead to reduced NCC activity. For approximately 10 percent of patients with a Gitelman syndrome phenotype, the genotype is unknown.
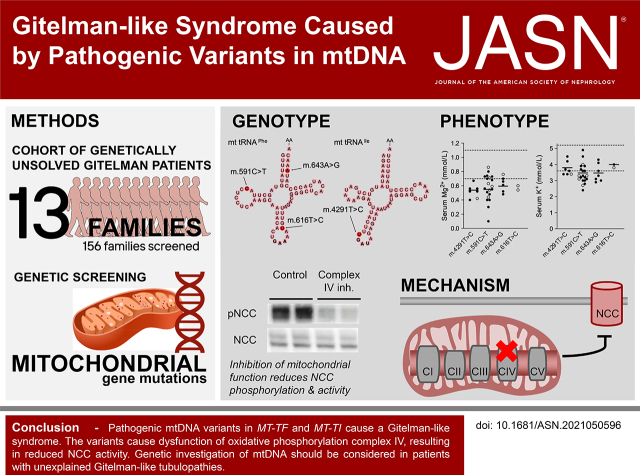
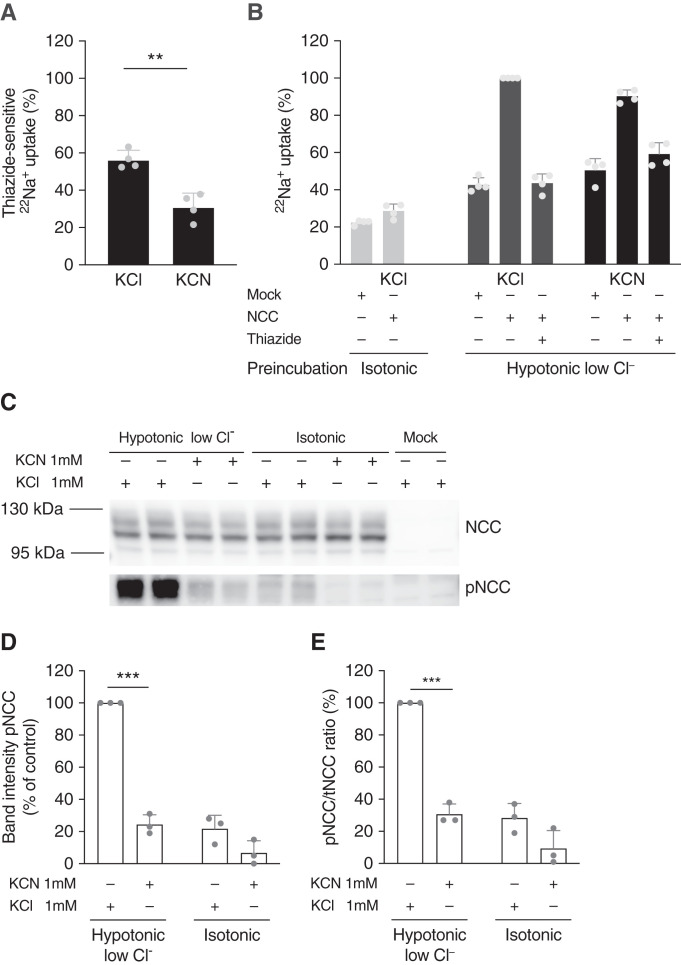
Methods: We identified mitochondrial DNA (mtDNA) variants in three families with Gitelman-like electrolyte abnormalities, then investigated 156 families for variants in MT-TI and MT-TF, which encode the transfer RNAs for phenylalanine and isoleucine. Mitochondrial respiratory chain function was assessed in patient fibroblasts. Mitochondrial dysfunction was induced in NCC-expressing HEK293 cells to assess the effect on thiazide-sensitive 22Na+ transport.
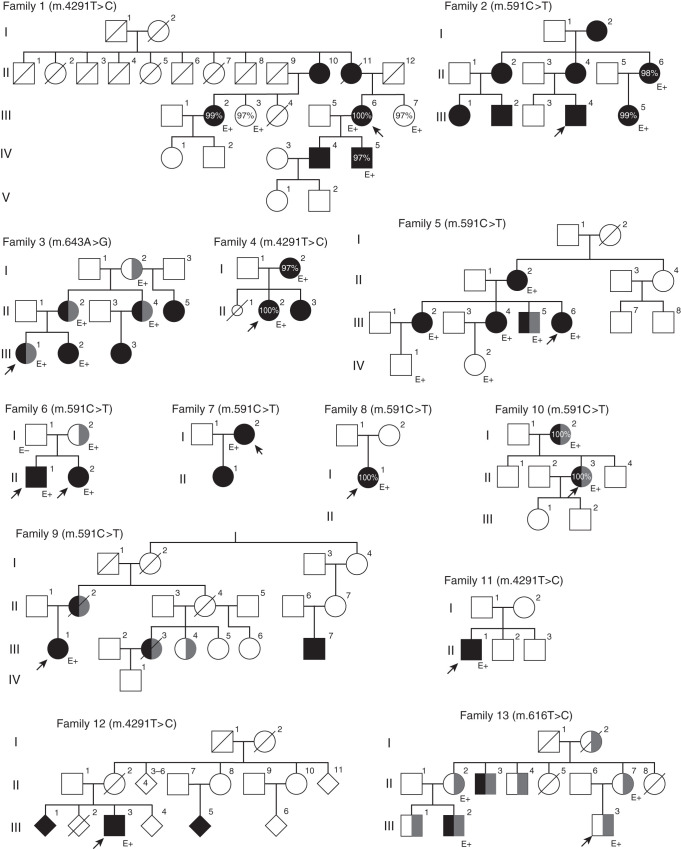
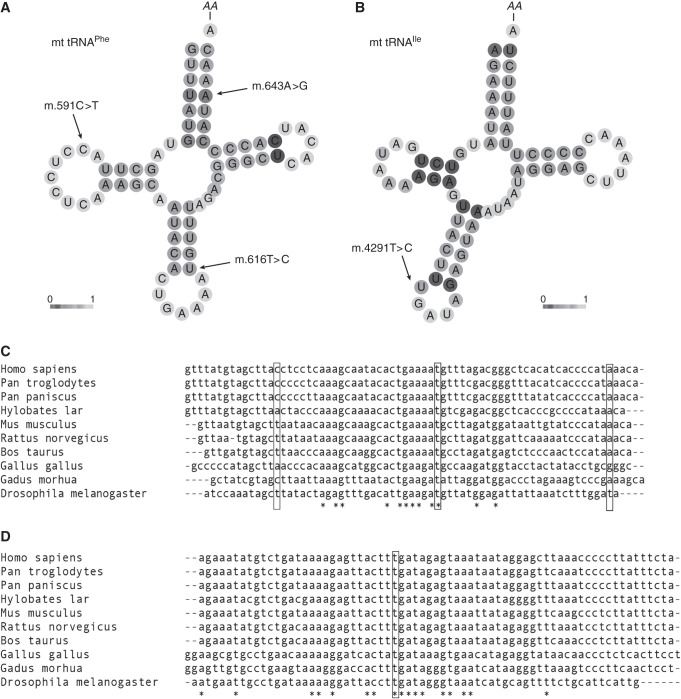
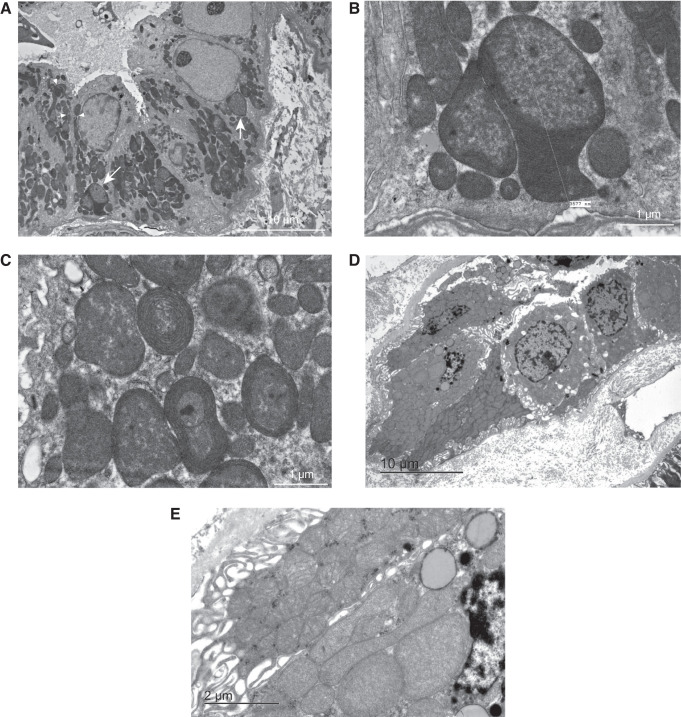
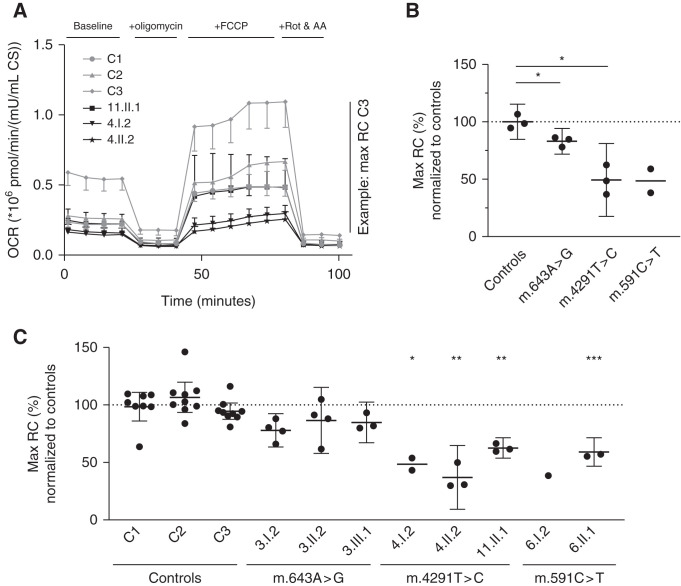
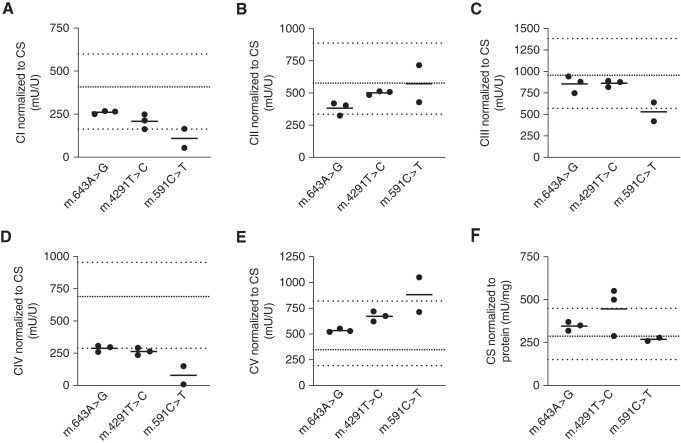
Results: Genetic investigations revealed four mtDNA variants in 13 families: m.591C>T (n=7), m.616T>C (n=1), m.643A>G (n=1) (all in MT-TF), and m.4291T>C (n=4, in MT-TI). Variants were near homoplasmic in affected individuals. All variants were classified as pathogenic, except for m.643A>G, which was classified as a variant of uncertain significance. Importantly, affected members of six families with an MT-TF variant additionally suffered from progressive chronic kidney disease. Dysfunction of oxidative phosphorylation complex IV and reduced maximal mitochondrial respiratory capacity were found in patient fibroblasts. In vitro pharmacological inhibition of complex IV, mimicking the effect of the mtDNA variants, inhibited NCC phosphorylation and NCC-mediated sodium uptake.
Conclusion: Pathogenic mtDNA variants in MT-TF and MT-TI can cause a Gitelman-like syndrome. Genetic investigation of mtDNA should be considered in patients with unexplained Gitelman syndrome-like tubulopathies.
Keywords: Gitelman-s syndrome; Na transport; blood pressure; chronic kidney disease; chronic kidney failure; epithelial sodium transport; genetic renal disease; human genetics; ion transport; mitochondria.
Copyright © 2022 by the American Society of Nephrology.
Figures
Comment in
-
mtDNA variants in Gitelman-like syndrome.Nat Rev Nephrol. 2021 Dec;17(12):794. doi: 10.1038/s41581-021-00508-1. Nat Rev Nephrol. 2021. PMID: 34663984 No abstract available.
References
-
- Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, et al. : Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996 - PubMed
-
- Blanchard A, Bockenhauer D, Bolignano D, Calò LA, Cosyns E, Devuyst O, et al. : Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 91: 24–33, 2017 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
