

Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms
- PMID: 34608321
- PMCID: PMC8793939
- DOI: 10.1038/s41592-021-01283-4
Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms
Abstract
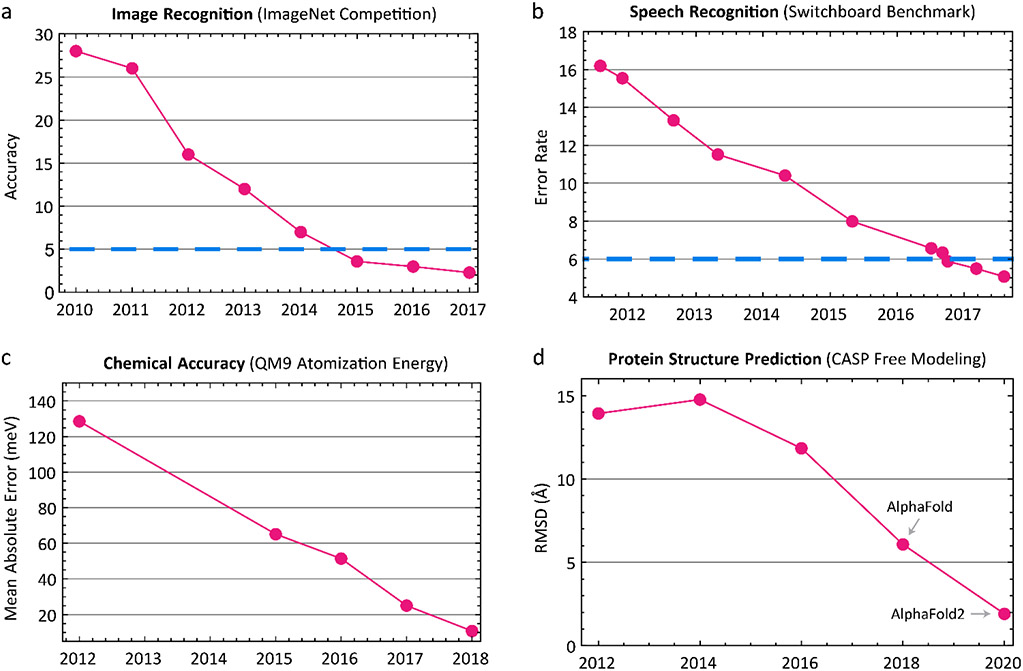
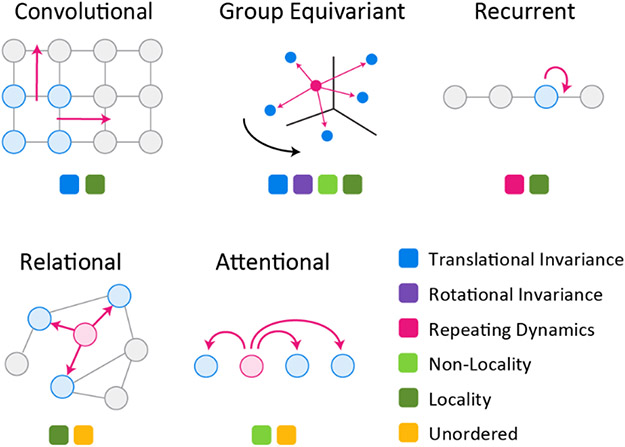
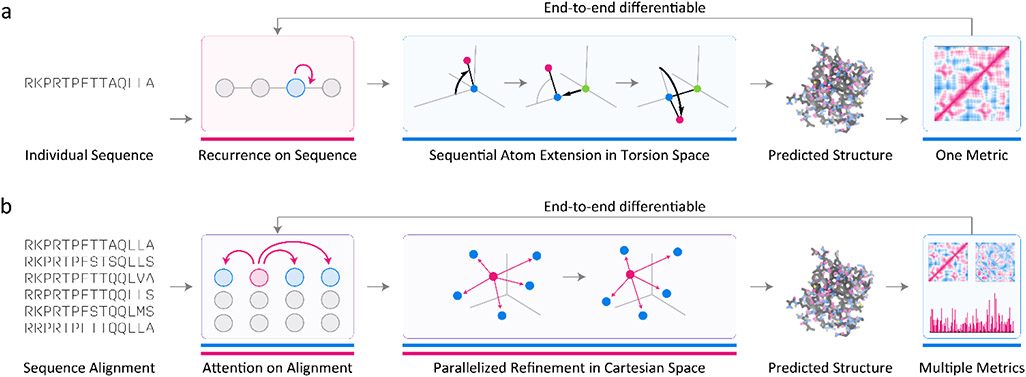
Deep learning using neural networks relies on a class of machine-learnable models constructed using 'differentiable programs'. These programs can combine mathematical equations specific to a particular domain of natural science with general-purpose, machine-learnable components trained on experimental data. Such programs are having a growing impact on molecular and cellular biology. In this Perspective, we describe an emerging 'differentiable biology' in which phenomena ranging from the small and specific (for example, one experimental assay) to the broad and complex (for example, protein folding) can be modeled effectively and efficiently, often by exploiting knowledge about basic natural phenomena to overcome the limitations of sparse, incomplete and noisy data. By distilling differentiable biology into a small set of conceptual primitives and illustrative vignettes, we show how it can help to address long-standing challenges in integrating multimodal data from diverse experiments across biological scales. This promises to benefit fields as diverse as biophysics and functional genomics.
© 2021. Springer Nature America, Inc.
Figures


References
-
- Abadi Martín et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems. http://tensorflow.org/ (2015).
-
- Paszke A et al. Automatic differentiation in PyTorch. (2017).
-
- Bradbury James, Frostig Roy, Hawkins Peter, Matthew James Johnson. JAX: Autograd and XLA. (Google, 2021).
-
- LeCun Y, Bengio Y & Hinton G Deep learning. Nature 521, 436–444 (2015). - PubMed
-
- He K, Zhang X, Ren S & Sun J Deep Residual Learning for Image Recognition. ArXiv151203385 Cs (2015).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
