

Effective gene expression prediction from sequence by integrating long-range interactions
- PMID: 34608324
- PMCID: PMC8490152
- DOI: 10.1038/s41592-021-01252-x
Effective gene expression prediction from sequence by integrating long-range interactions
Abstract
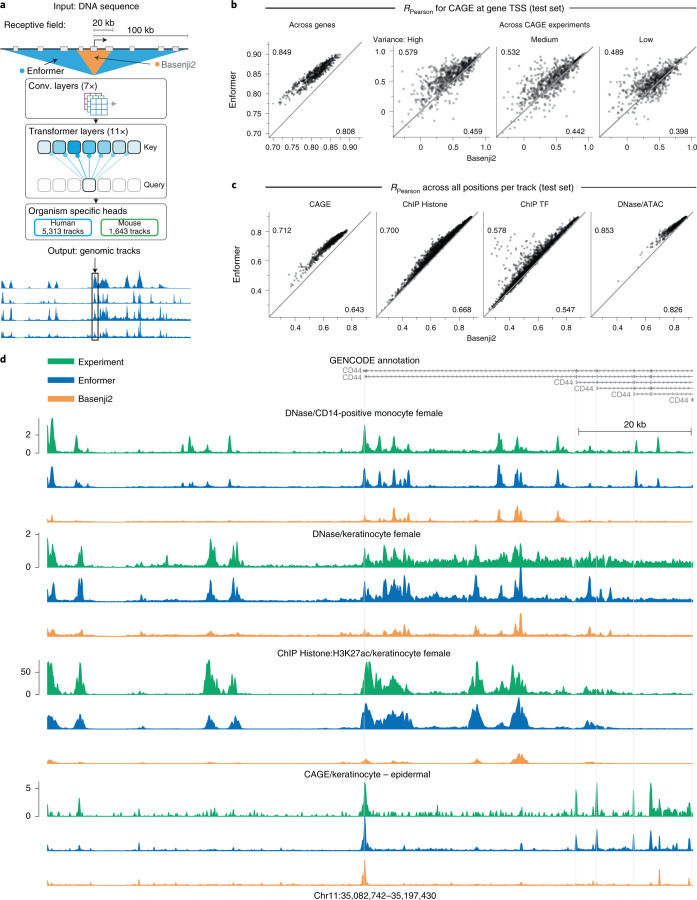
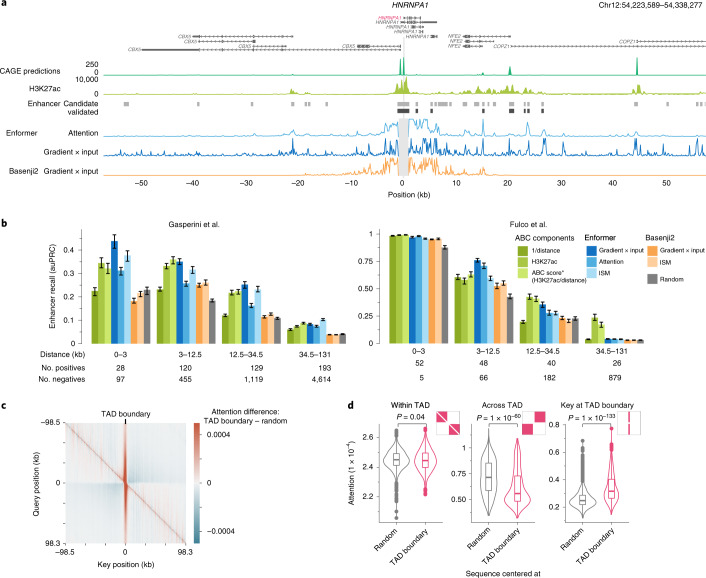
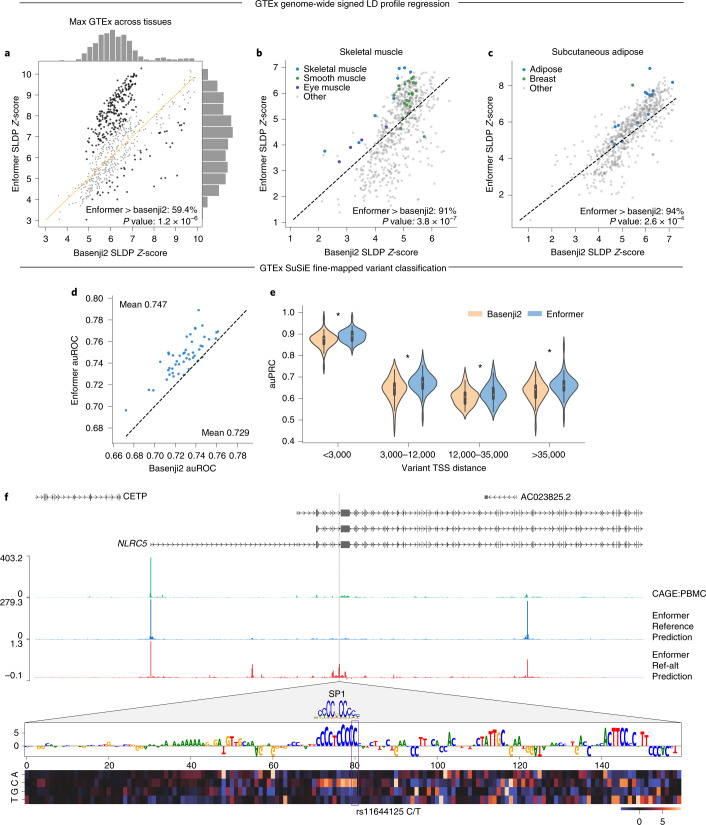
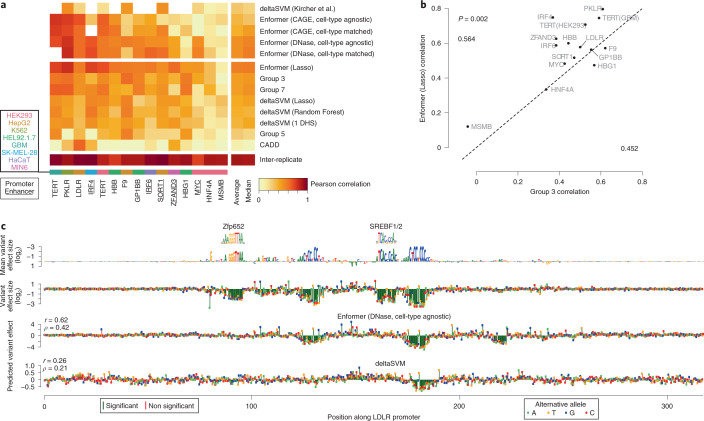
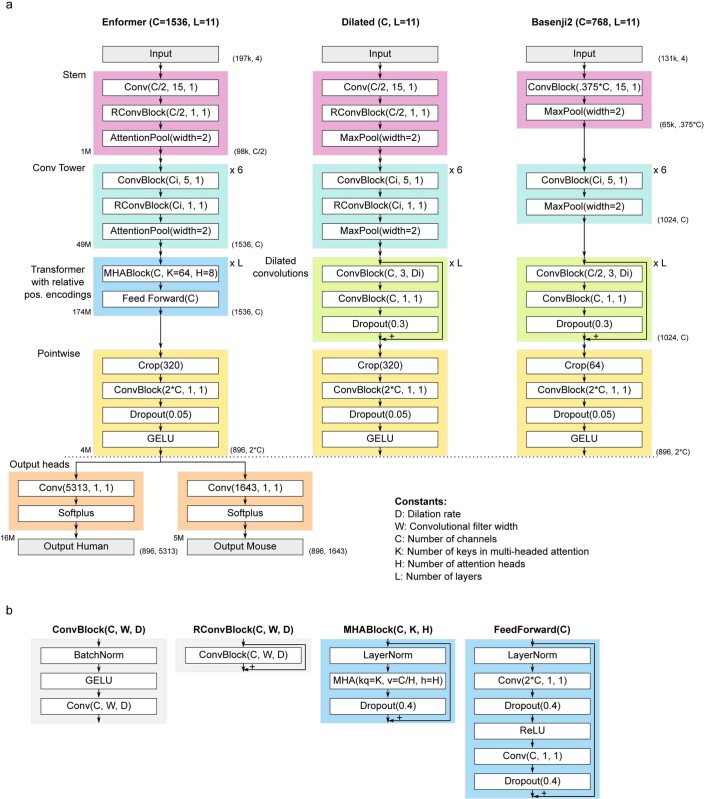
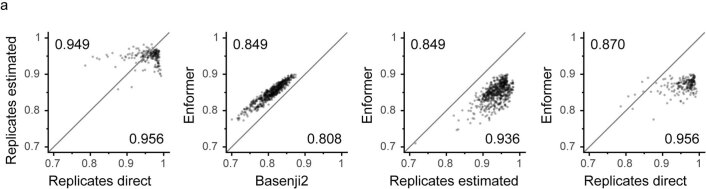
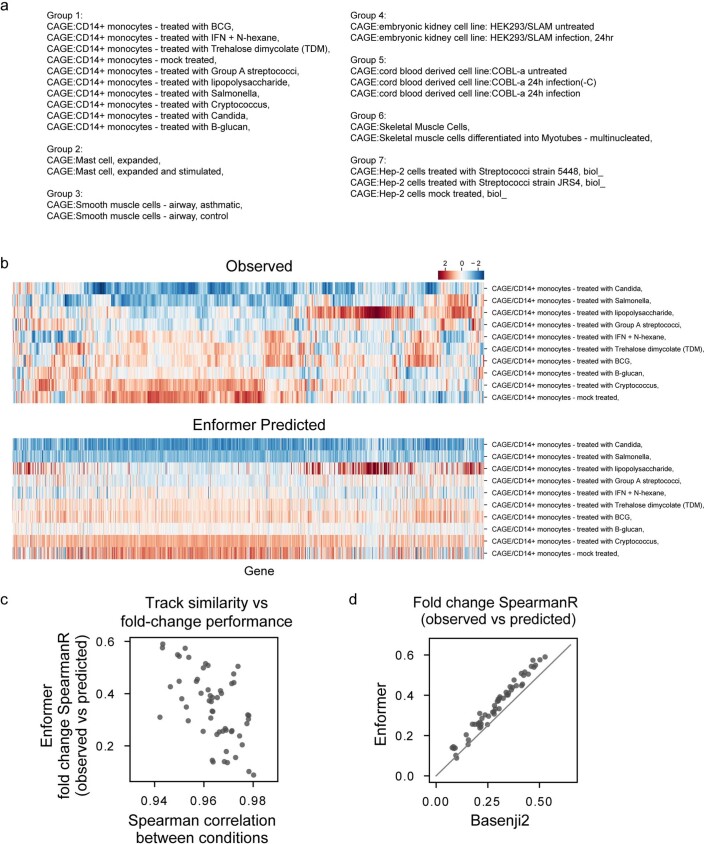
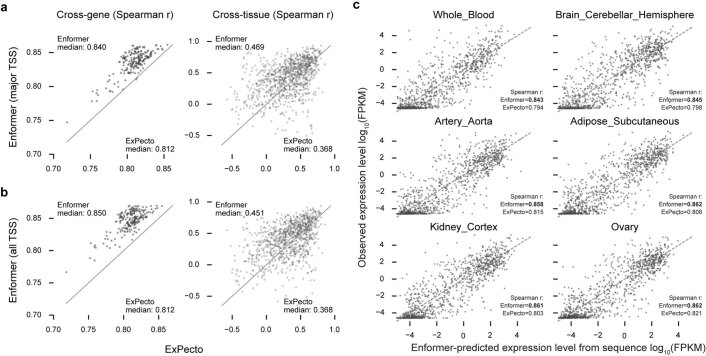
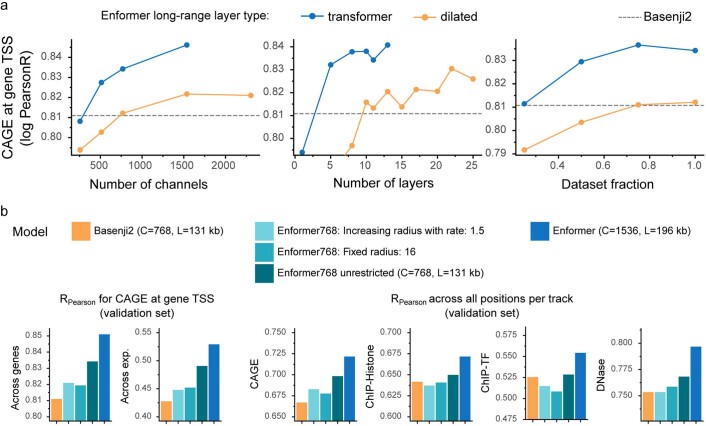
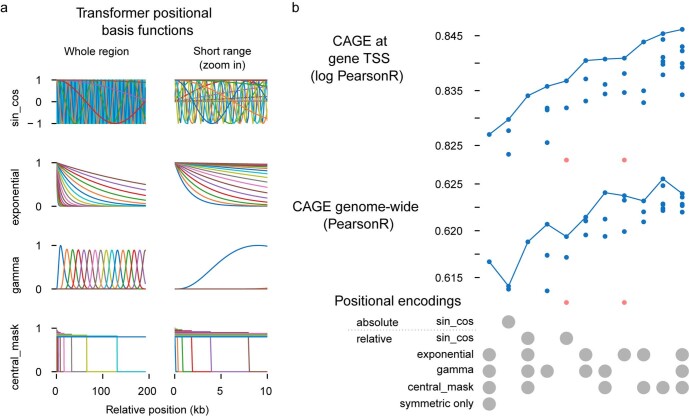
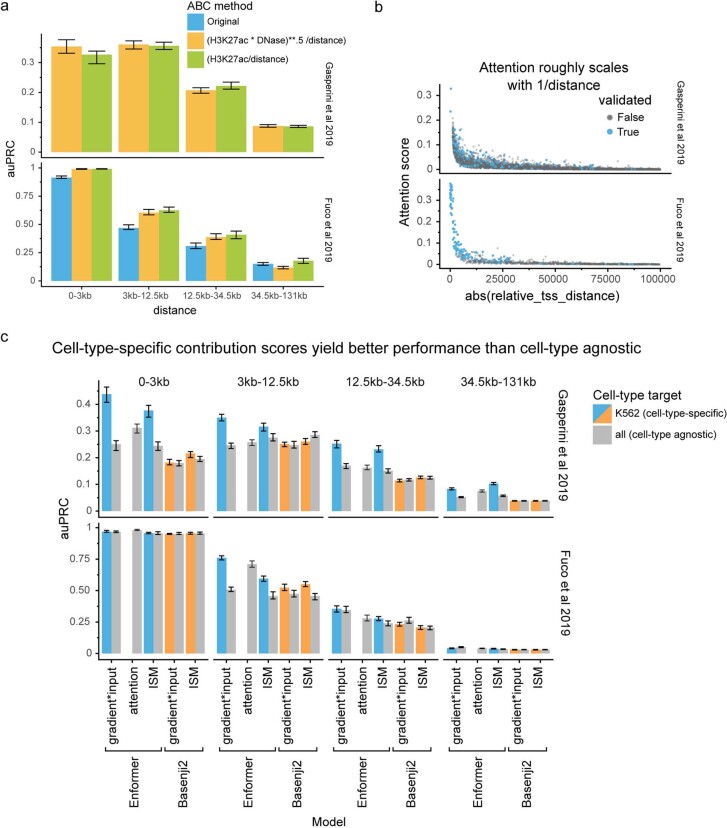
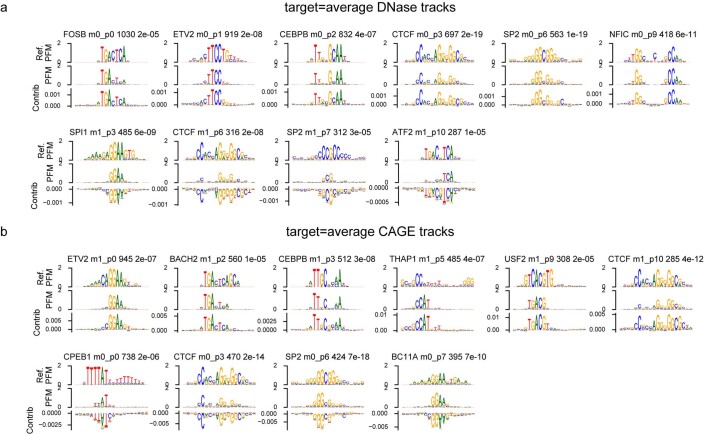
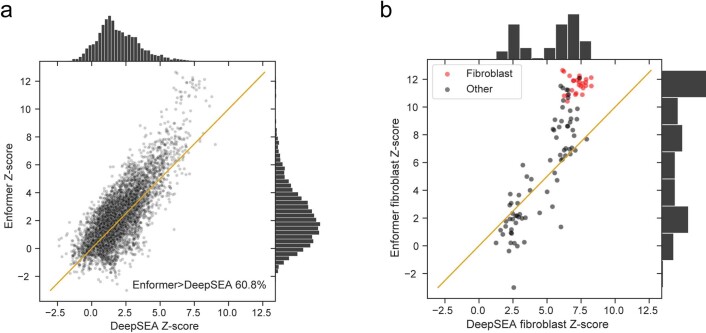
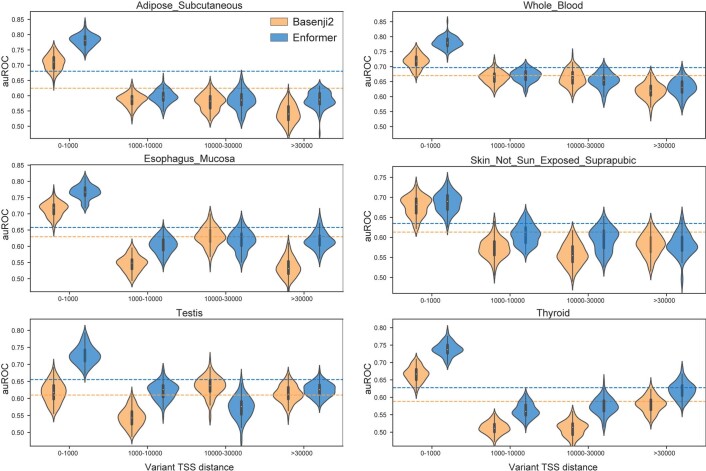
How noncoding DNA determines gene expression in different cell types is a major unsolved problem, and critical downstream applications in human genetics depend on improved solutions. Here, we report substantially improved gene expression prediction accuracy from DNA sequences through the use of a deep learning architecture, called Enformer, that is able to integrate information from long-range interactions (up to 100 kb away) in the genome. This improvement yielded more accurate variant effect predictions on gene expression for both natural genetic variants and saturation mutagenesis measured by massively parallel reporter assays. Furthermore, Enformer learned to predict enhancer-promoter interactions directly from the DNA sequence competitively with methods that take direct experimental data as input. We expect that these advances will enable more effective fine-mapping of human disease associations and provide a framework to interpret cis-regulatory evolution.
© 2021. The Author(s).
Conflict of interest statement
Ž.A., A.G-B., K.R.T., Y.A., J.J., and P.K. are employed by DeepMind. V.A., and D.R.K. are employed by Calico Life Sciences. J.R.L. is employed by Google. The remaining authors declare no competing interests.
Figures
Comment in
-
A wider field of view to predict expression.Nat Methods. 2021 Oct;18(10):1155-1156. doi: 10.1038/s41592-021-01259-4. Nat Methods. 2021. PMID: 34608323 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
