

Targeting cholesterol homeostasis in hematopoietic malignancies
- PMID: 34610110
- PMCID: PMC8814816
- DOI: 10.1182/blood.2021012788
Targeting cholesterol homeostasis in hematopoietic malignancies
Abstract
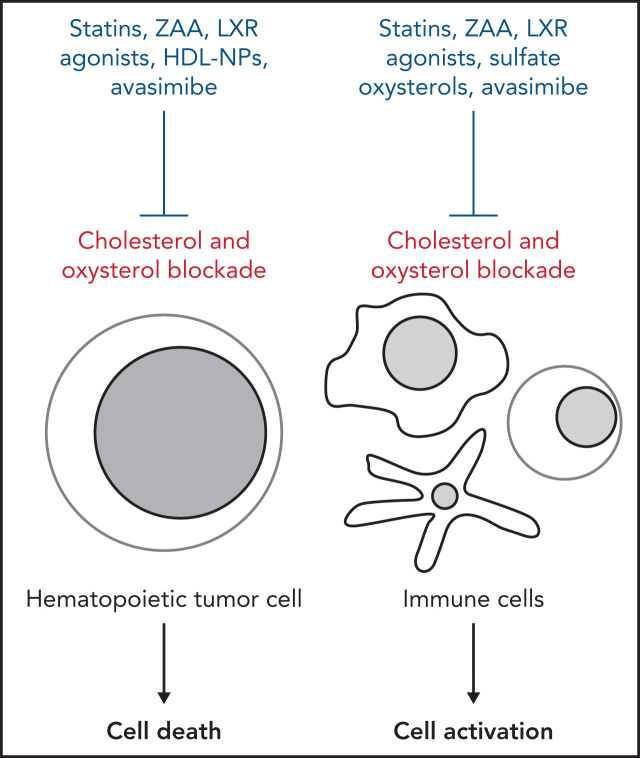
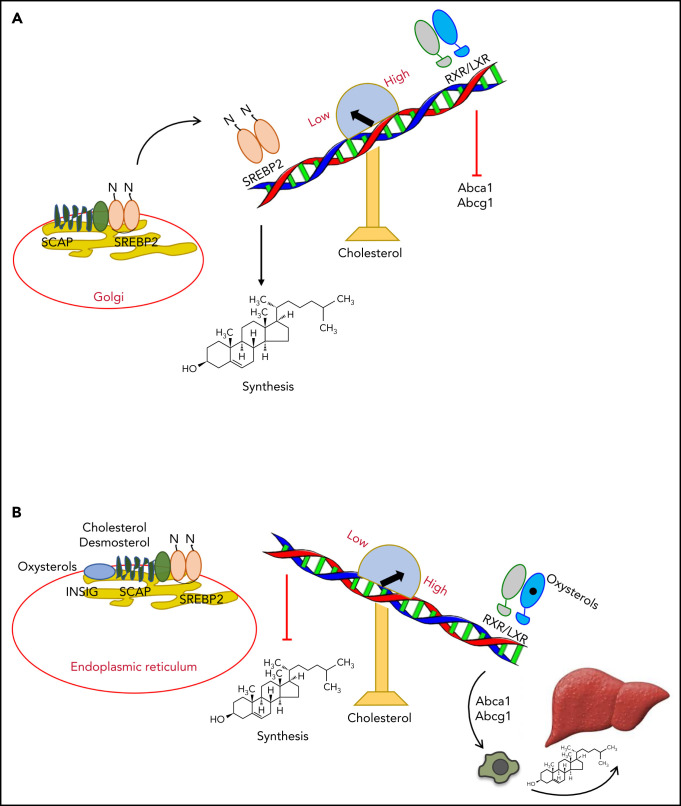
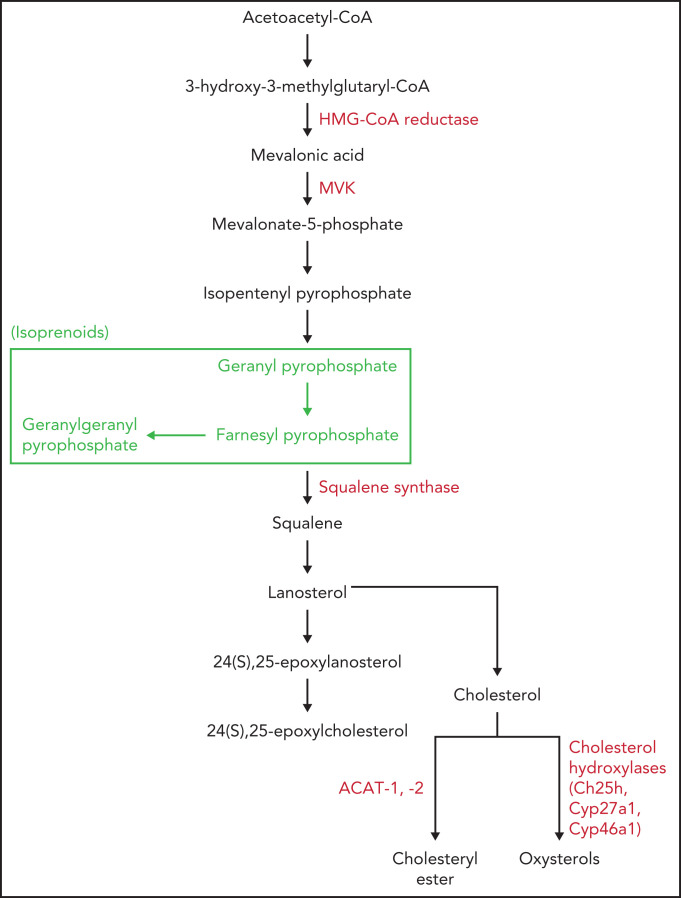
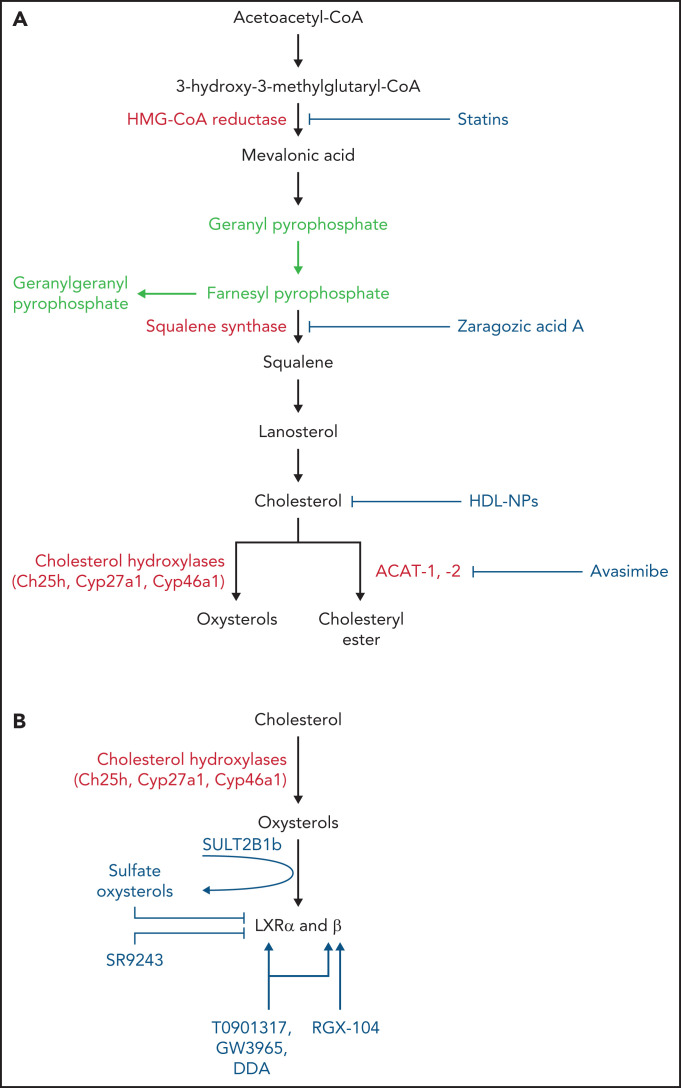
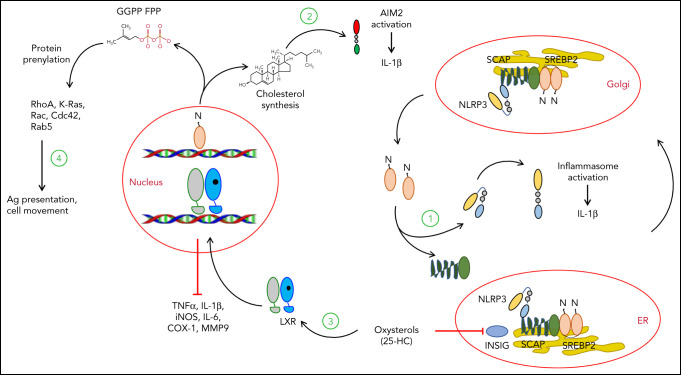
Cholesterol is a vital lipid for cellular functions. It is necessary for membrane biogenesis, cell proliferation, and differentiation. In addition to maintaining cell integrity and permeability, increasing evidence indicates a strict link between cholesterol homeostasis, inflammation, and hematological tumors. This makes cholesterol homeostasis an optimal therapeutic target for hematopoietic malignancies. Manipulating cholesterol homeostasis by either interfering with its synthesis or activating the reverse cholesterol transport via the engagement of liver X receptors affects the integrity of tumor cells both in vitro and in vivo. Cholesterol homeostasis has also been manipulated to restore antitumor immune responses in preclinical models. These observations have prompted clinical trials involving acute myeloid leukemia to test the combination of chemotherapy with drugs interfering with cholesterol synthesis (ie, statins). We review the role of cholesterol homeostasis in hematopoietic malignancies as well as in cells of the tumor microenvironment and discuss the potential use of lipid modulators for therapeutic purposes.
© 2022 by The American Society of Hematology.
Figures
References
-
- Brown MS, Goldstein JL.. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997; 89(3):331-340. - PubMed
-
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ.. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383(6602): 728-731. - PubMed
-
- Goldstein JL, DeBose-Boyd RA, Brown MS.. Protein sensors for membrane sterols. Cell. 2006;124(1):35-46. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
