

Novel therapeutic targets for cholestatic and fatty liver disease
- PMID: 34615727
- PMCID: PMC8666813
- DOI: 10.1136/gutjnl-2021-324305
Novel therapeutic targets for cholestatic and fatty liver disease
Abstract
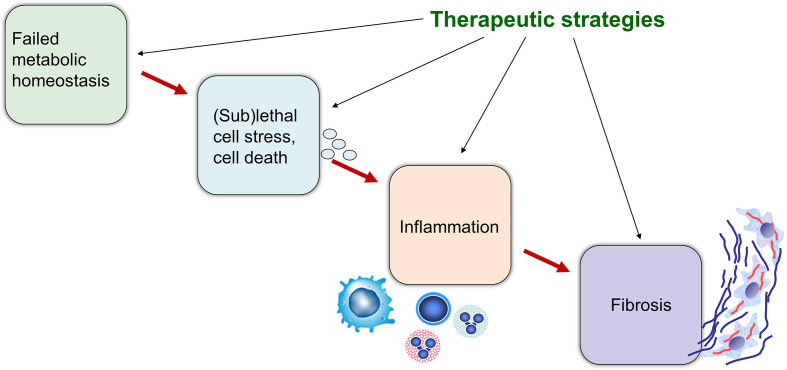
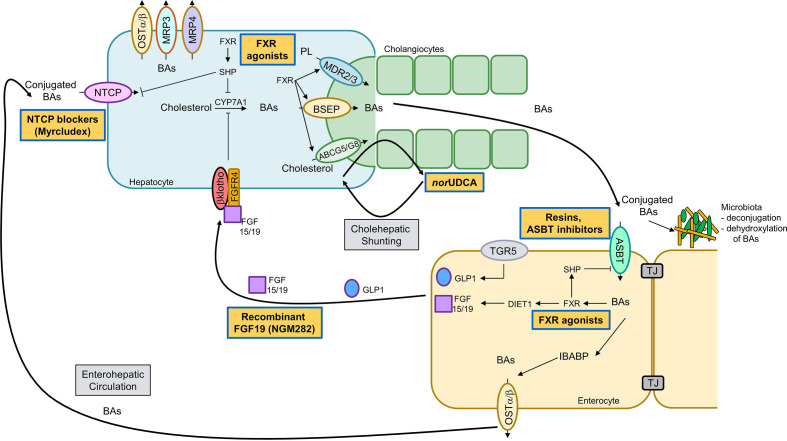
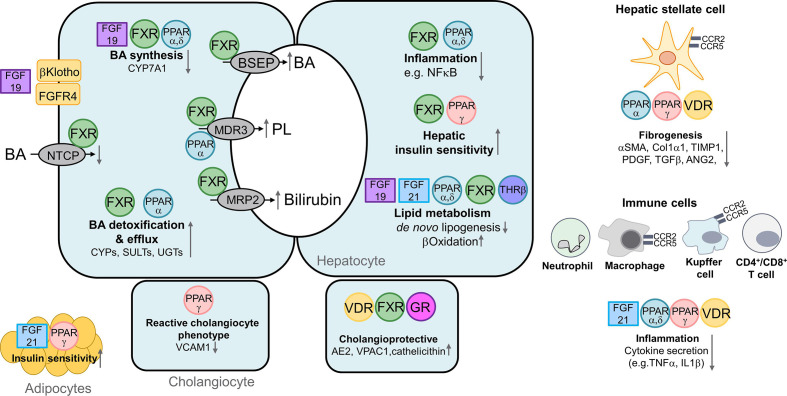
Cholestatic and non-alcoholic fatty liver disease (NAFLD) share several key pathophysiological mechanisms which can be targeted by novel therapeutic concepts that are currently developed for both areas. Nuclear receptors (NRs) are ligand-activated transcriptional regulators of key metabolic processes including hepatic lipid and glucose metabolism, energy expenditure and bile acid (BA) homoeostasis, as well as inflammation, fibrosis and cellular proliferation. Dysregulation of these processes contributes to the pathogenesis and progression of cholestatic as well as fatty liver disease, placing NRs at the forefront of novel therapeutic approaches. This includes BA and fatty acid activated NRs such as farnesoid-X receptor (FXR) and peroxisome proliferator-activated receptors, respectively, for which high affinity therapeutic ligands targeting specific or multiple isoforms have been developed. Moreover, novel liver-specific ligands for thyroid hormone receptor beta 1 complete the spectrum of currently available NR-targeted drugs. Apart from FXR ligands, BA signalling can be targeted by mimetics of FXR-activated fibroblast growth factor 19, modulation of their enterohepatic circulation through uptake inhibitors in hepatocytes and enterocytes, as well as novel BA derivatives undergoing cholehepatic shunting (instead of enterohepatic circulation). Other therapeutic approaches more directly target inflammation and/or fibrosis as critical events of disease progression. Combination strategies synergistically targeting metabolic disturbances, inflammation and fibrosis may be ultimately necessary for successful treatment of these complex and multifactorial disorders.
Keywords: fibrosis; inflammation.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: MT: Consulting: Albireo, BiomX, Boehringer Ingelheim, Falk, Genfit, Intercept, Janssen, MSD, Gilead, Novartis, Shire, Phenex, Regulus. Speakers bureau: Falk Foundation, Gilad, Intercept, MSD. Grants: Albireo, Cymabay, Falk, Gilead, Intercept, MSD, Takeda, Alnylam, Ultragenyx. Travel grants: Abbvie, Falk, Gilead, Intercept. Intellectual property rights: Co Inventor or Patent on Medical Use nor UDCA. CDF received travel grants from Falk and Gilead.
Figures
Comment in
-
FGF-21 analogues for treatment of non-alcoholic steatohepatitis and fibrosis: a meta-analysis with fragility index of phase 2 randomised placebo-controlled trials.Gut. 2024 Jul 11;73(8):1400-1402. doi: 10.1136/gutjnl-2023-331115. Gut. 2024. PMID: 37758327 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical