

The GPR171 pathway suppresses T cell activation and limits antitumor immunity
- PMID: 34615877
- PMCID: PMC8494883
- DOI: 10.1038/s41467-021-26135-9
The GPR171 pathway suppresses T cell activation and limits antitumor immunity
Abstract
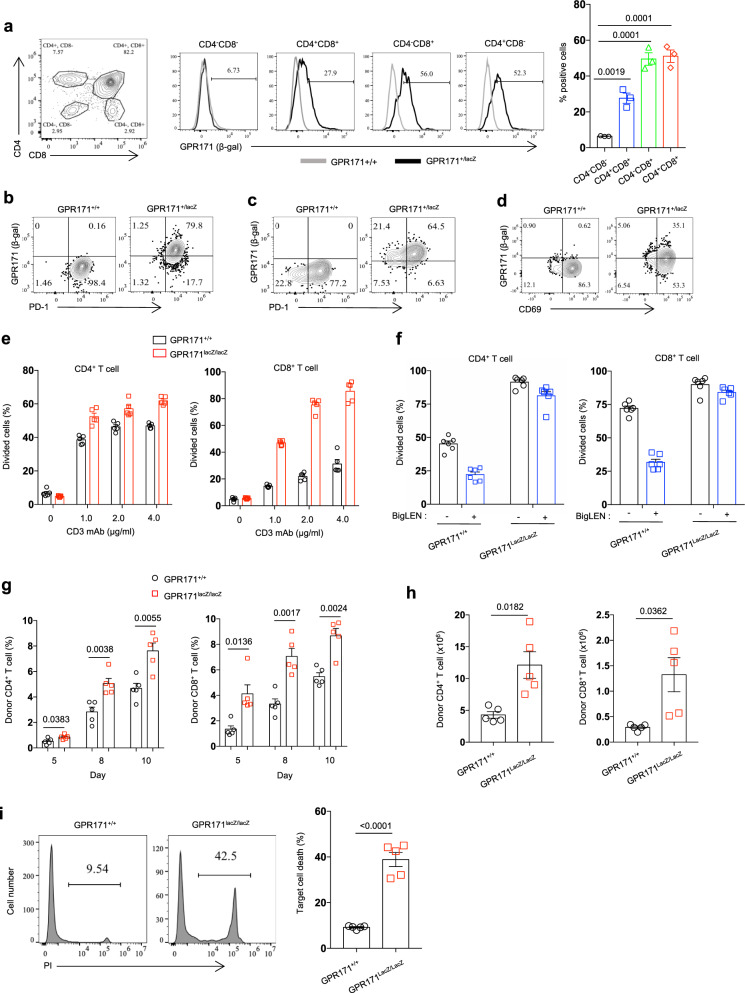
The recently identified G-protein-coupled receptor GPR171 and its ligand BigLEN are thought to regulate food uptake and anxiety. Though GPR171 is commonly used as a T cell signature gene in transcriptomic studies, its potential role in T cell immunity has not been explored. Here we show that GPR171 is transcribed in T cells and its protein expression is induced upon antigen stimulation. The neuropeptide ligand BigLEN interacts with GPR171 to suppress T cell receptor-mediated signalling pathways and to inhibit T cell proliferation. Loss of GPR171 in T cells leads to hyperactivity to antigen stimulation and GPR171 knockout mice exhibit enhanced antitumor immunity. Blockade of GPR171 signalling by an antagonist promotes antitumor T cell immunity and improves immune checkpoint blockade therapies. Together, our study identifies the GPR171/BigLEN axis as a T cell checkpoint pathway that can be modulated for cancer immunotherapy.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
