

Pig genome functional annotation enhances the biological interpretation of complex traits and human disease
- PMID: 34615879
- PMCID: PMC8494738
- DOI: 10.1038/s41467-021-26153-7
Pig genome functional annotation enhances the biological interpretation of complex traits and human disease
Abstract
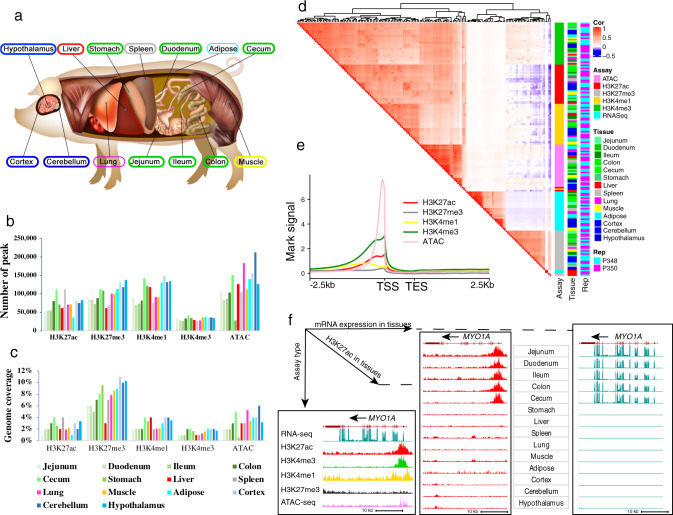
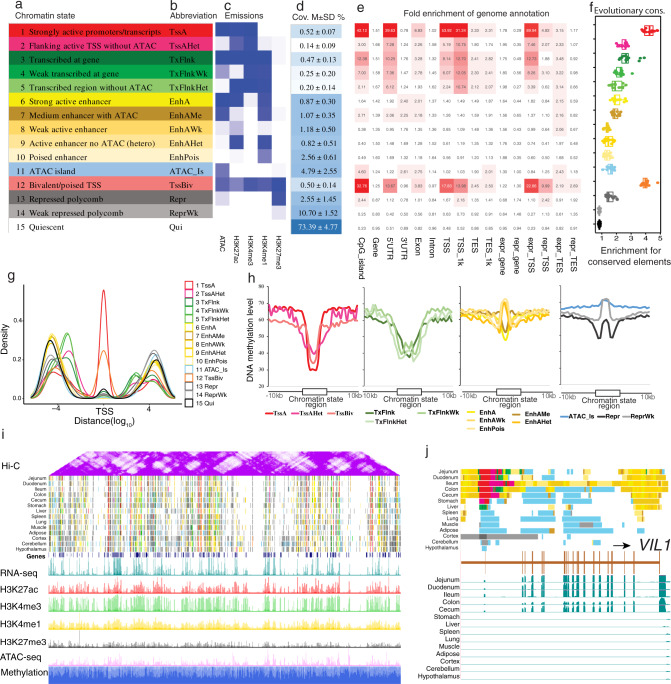
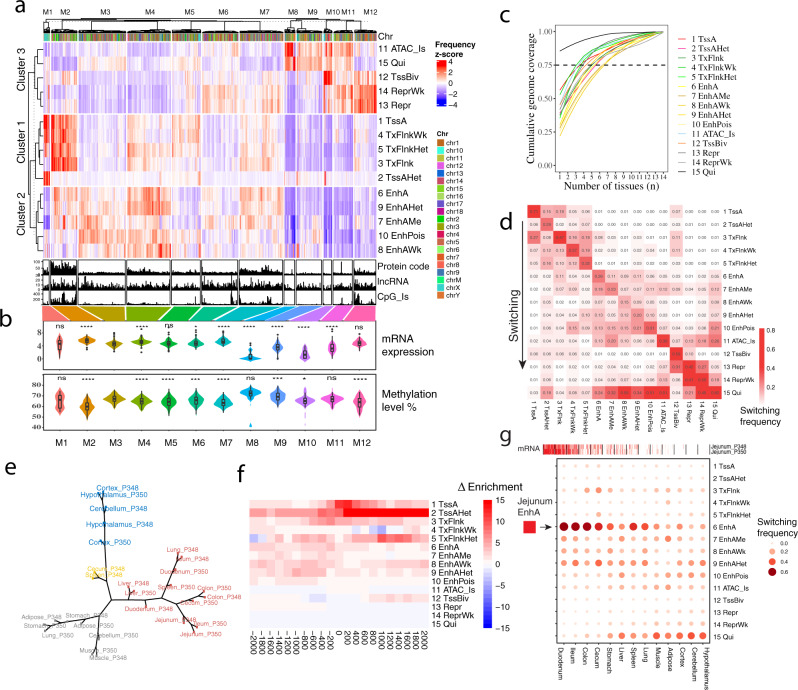
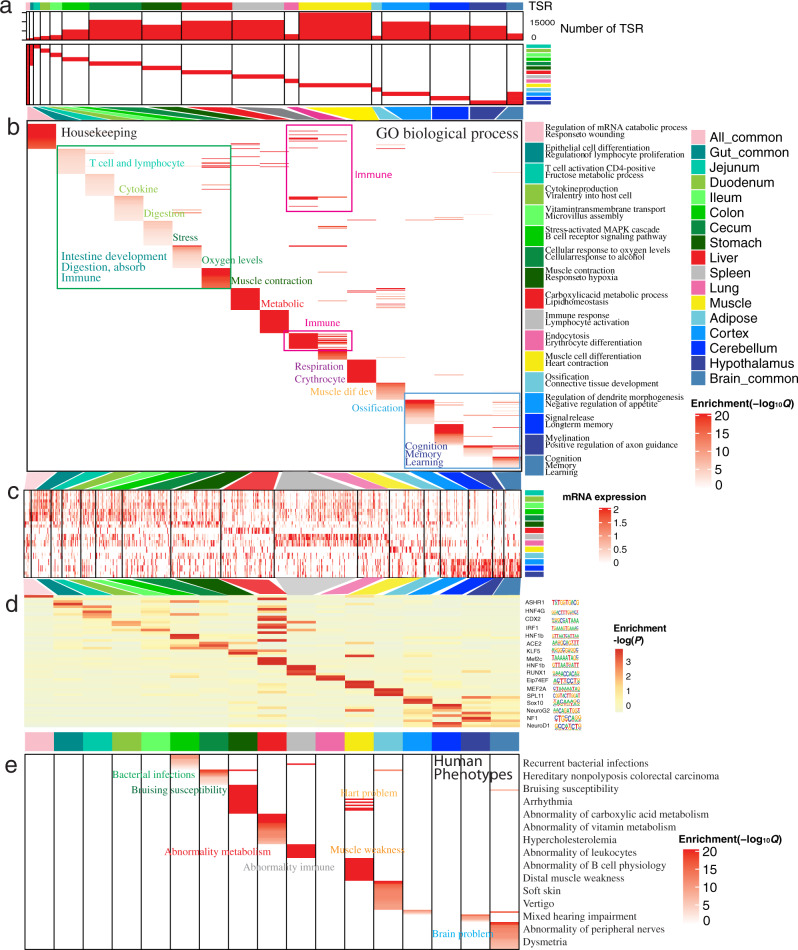
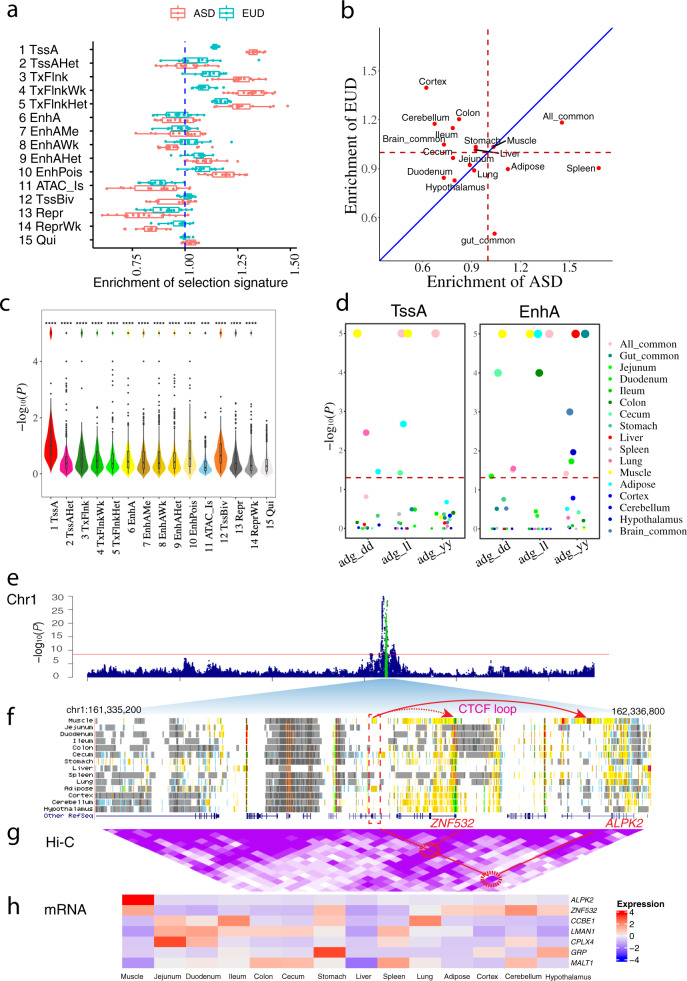
The functional annotation of livestock genomes is crucial for understanding the molecular mechanisms that underpin complex traits of economic importance, adaptive evolution and comparative genomics. Here, we provide the most comprehensive catalogue to date of regulatory elements in the pig (Sus scrofa) by integrating 223 epigenomic and transcriptomic data sets, representing 14 biologically important tissues. We systematically describe the dynamic epigenetic landscape across tissues by functionally annotating 15 different chromatin states and defining their tissue-specific regulatory activities. We demonstrate that genomic variants associated with complex traits and adaptive evolution in pig are significantly enriched in active promoters and enhancers. Furthermore, we reveal distinct tissue-specific regulatory selection between Asian and European pig domestication processes. Compared with human and mouse epigenomes, we show that porcine regulatory elements are more conserved in DNA sequence, under both rapid and slow evolution, than those under neutral evolution across pig, mouse, and human. Finally, we provide biological insights on tissue-specific regulatory conservation, and by integrating 47 human genome-wide association studies, we demonstrate that, depending on the traits, mouse or pig might be more appropriate biomedical models for different complex traits and diseases.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
