

POLθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis
- PMID: 34616022
- PMCID: PMC8675436
- DOI: 10.1038/s41556-021-00764-0
POLθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis
Erratum in
-
Publisher Correction: POLθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis.Nat Cell Biol. 2022 Jan;24(1):124. doi: 10.1038/s41556-021-00797-5. Nat Cell Biol. 2022. PMID: 34707239 No abstract available.
Abstract
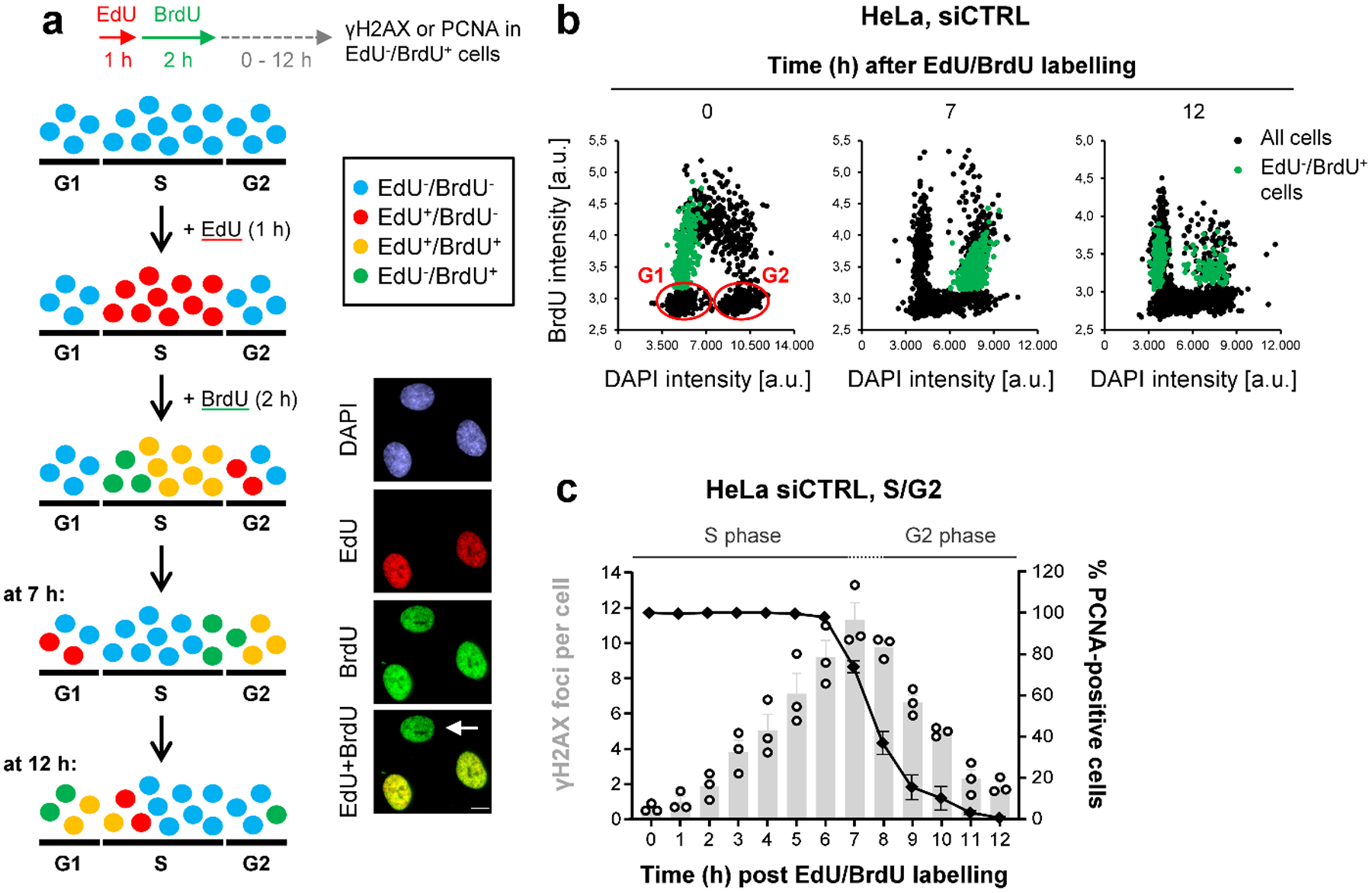
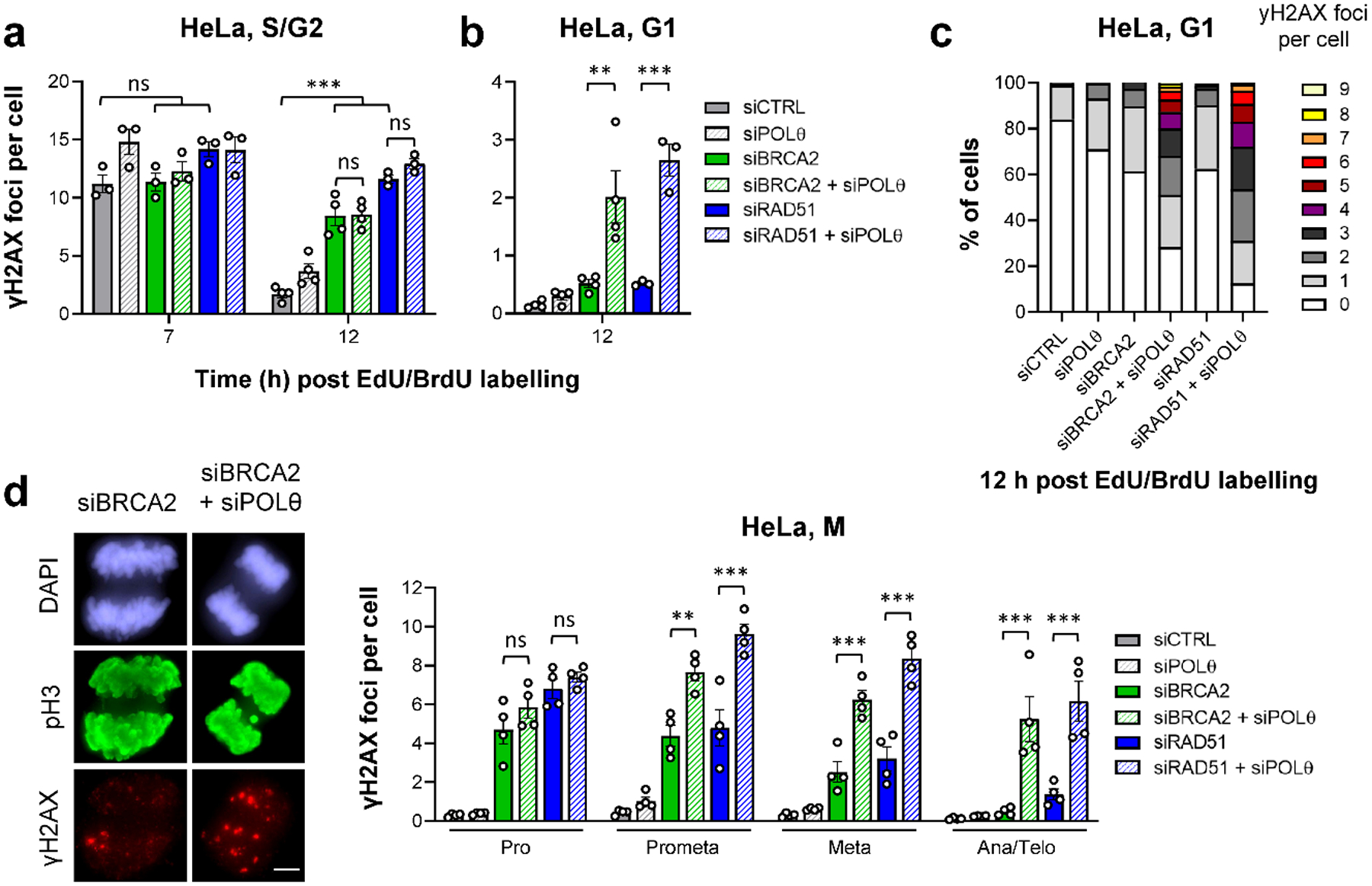
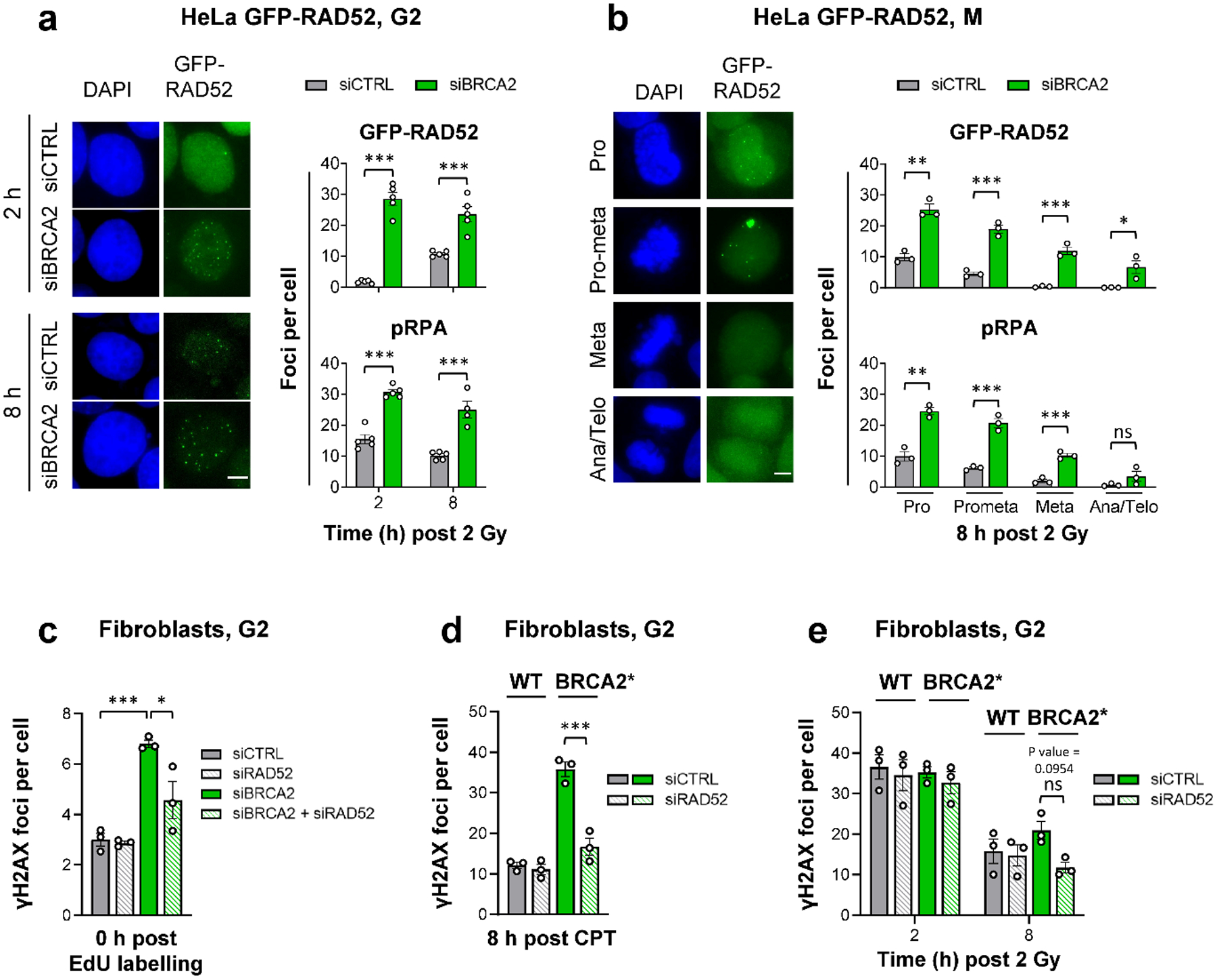
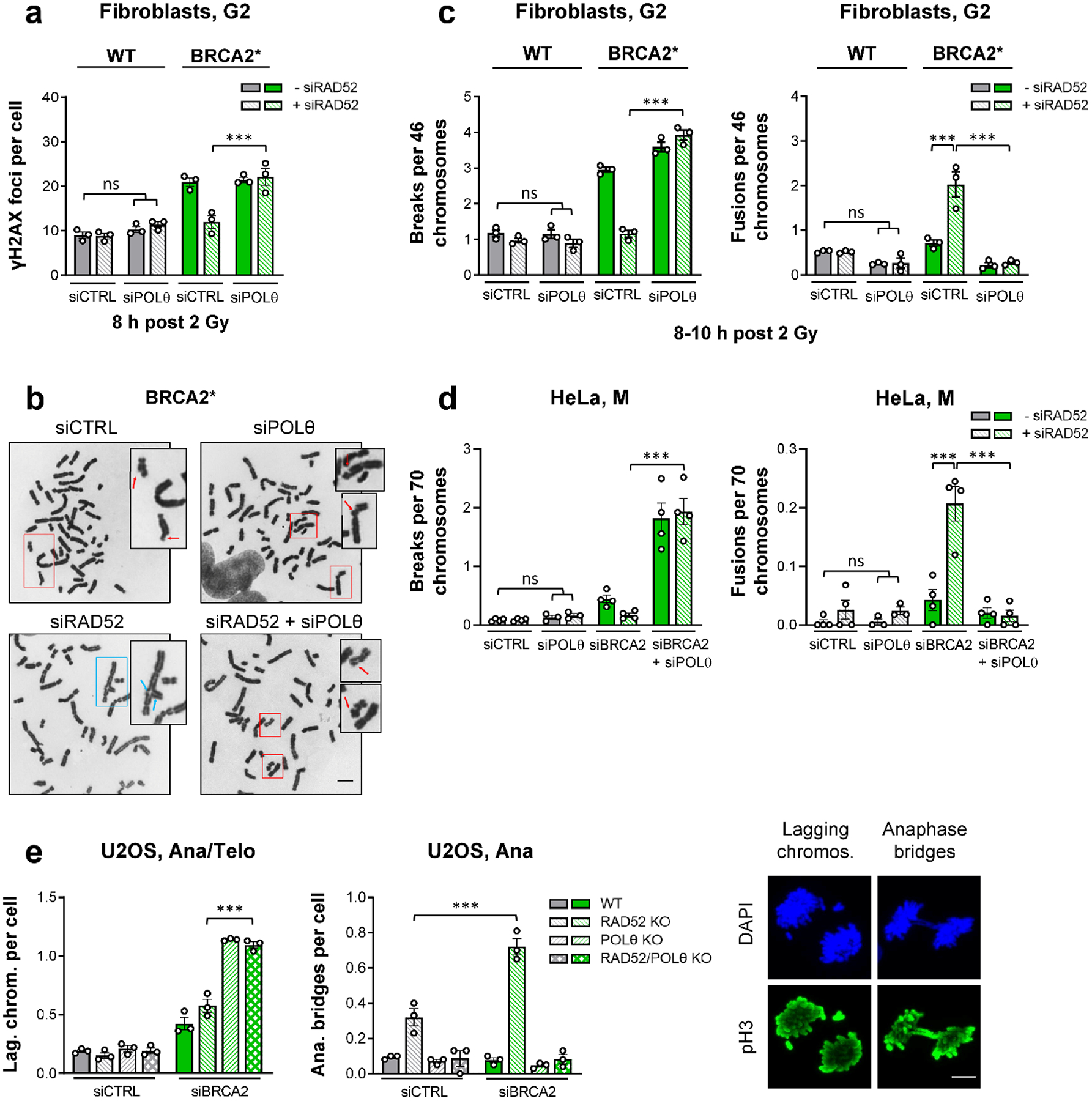
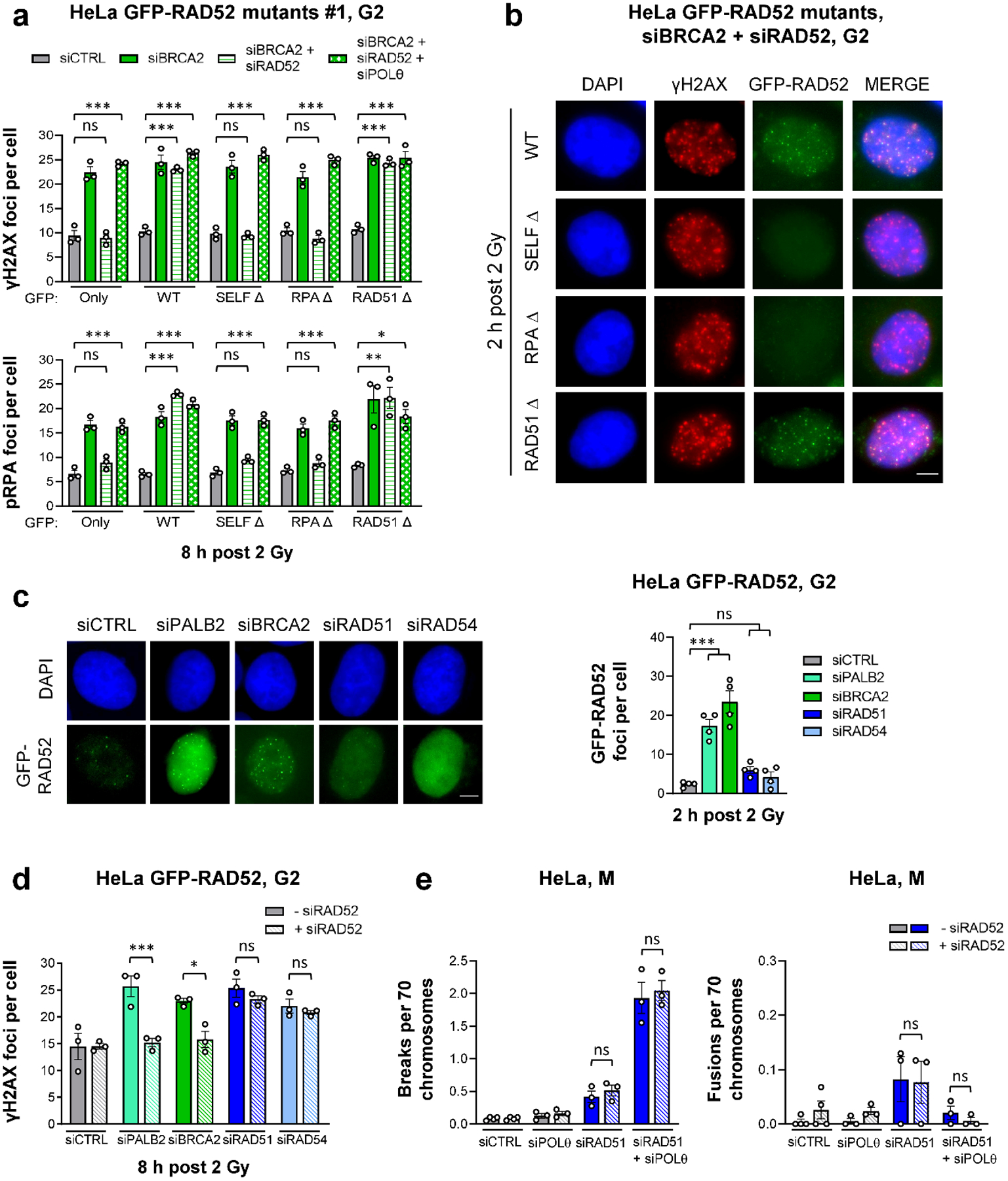
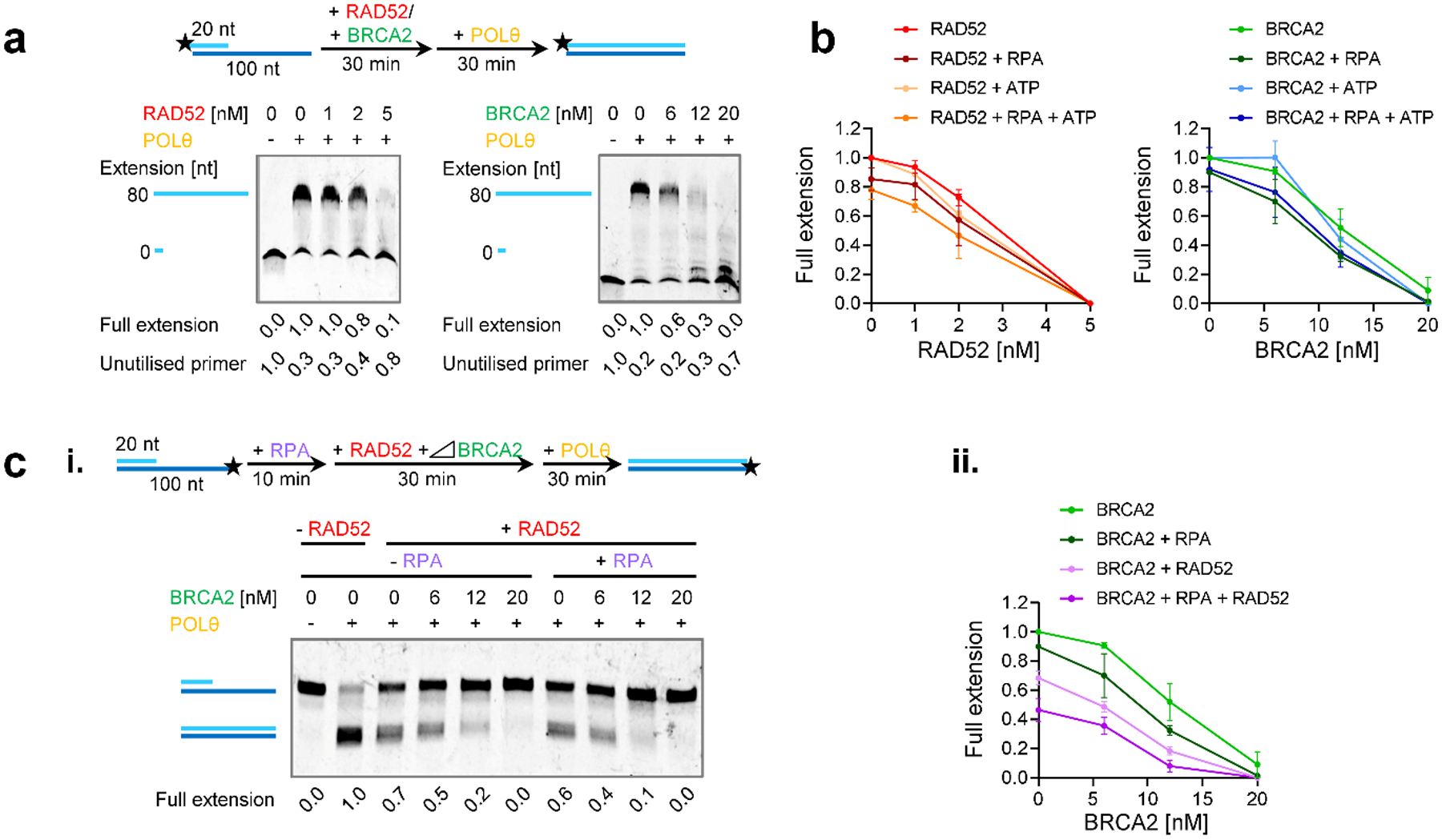
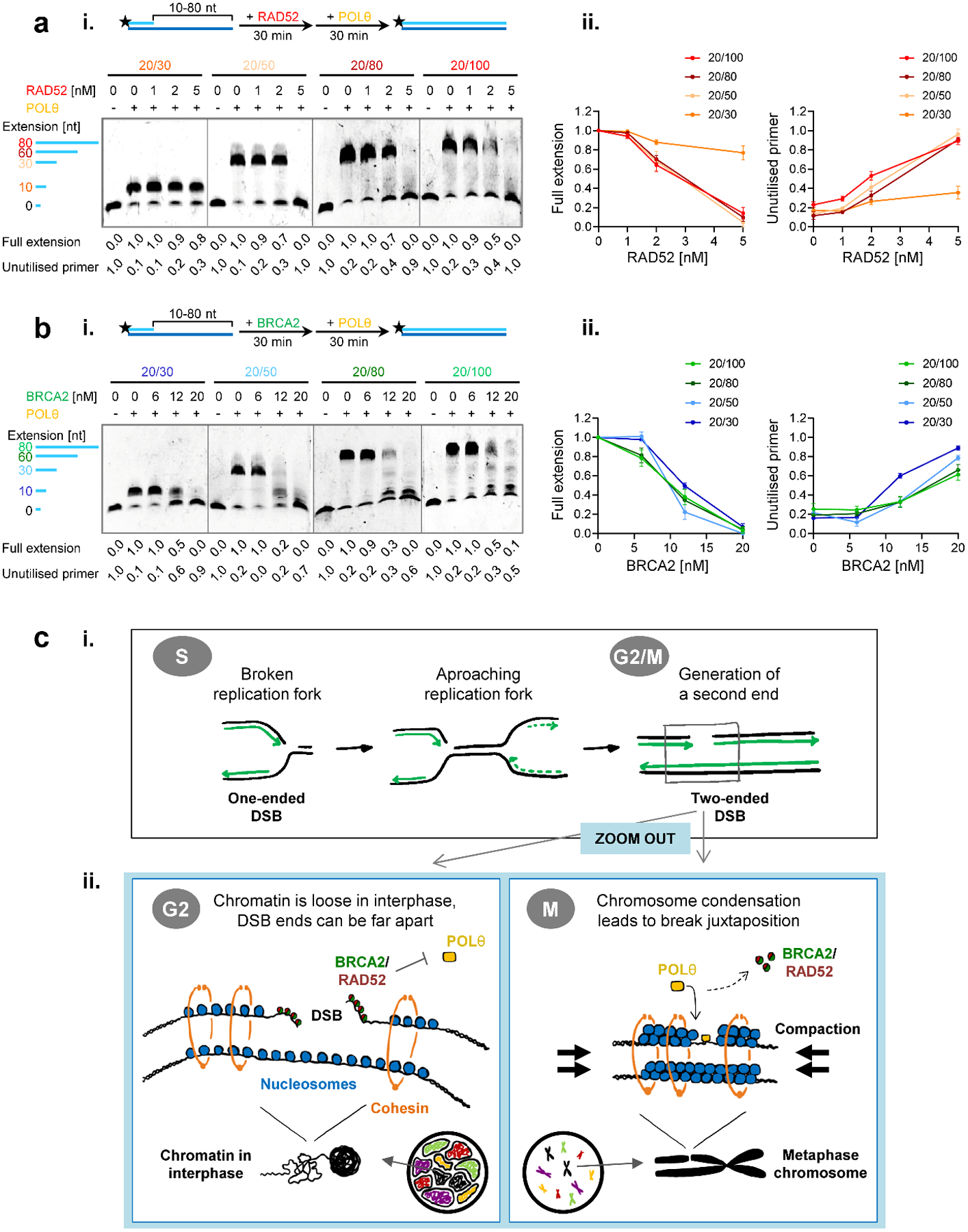
BRCA2-mutant cells are defective in homologous recombination, making them vulnerable to the inactivation of other pathways for the repair of DNA double-strand breaks (DSBs). This concept can be clinically exploited but is currently limited due to insufficient knowledge about how DSBs are repaired in the absence of BRCA2. We show that DNA polymerase θ (POLθ)-mediated end joining (TMEJ) repairs DSBs arising during the S phase in BRCA2-deficient cells only after the onset of the ensuing mitosis. This process is regulated by RAD52, whose loss causes the premature usage of TMEJ and the formation of chromosomal fusions. Purified RAD52 and BRCA2 proteins both block the DNA polymerase function of POLθ, suggesting a mechanism explaining their synthetic lethal relationships. We propose that the delay of TMEJ until mitosis ensures the conversion of originally one-ended DSBs into two-ended DSBs. Mitotic chromatin condensation might further serve to juxtapose correct break ends and limit chromosomal fusions.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
