

DNA methylation atlas of the mouse brain at single-cell resolution
- PMID: 34616061
- PMCID: PMC8494641
- DOI: 10.1038/s41586-020-03182-8
DNA methylation atlas of the mouse brain at single-cell resolution
Abstract
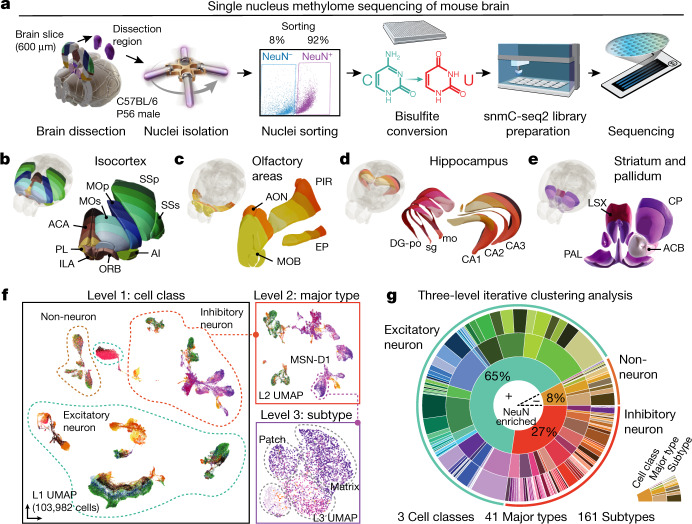
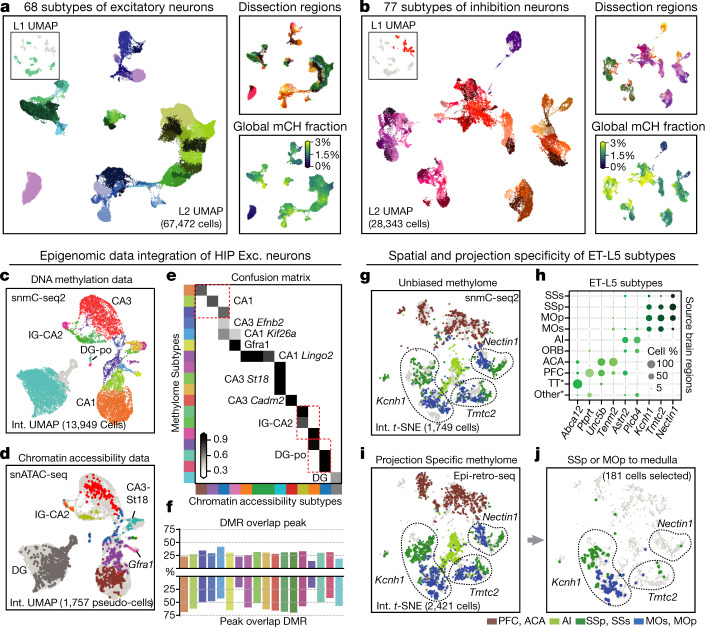
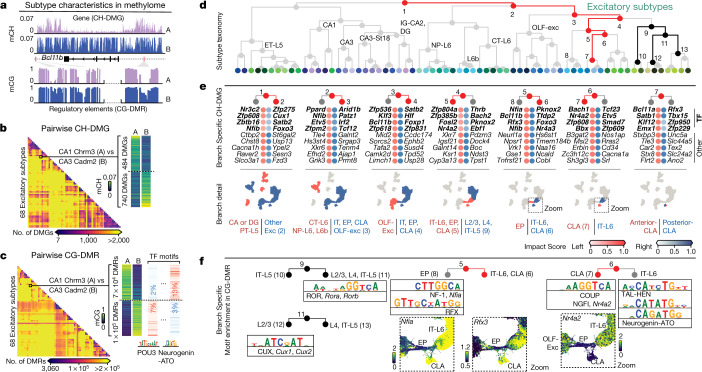
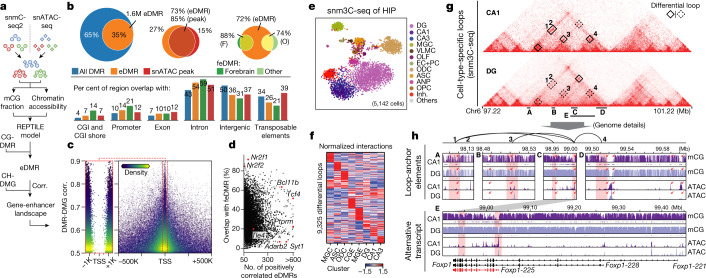
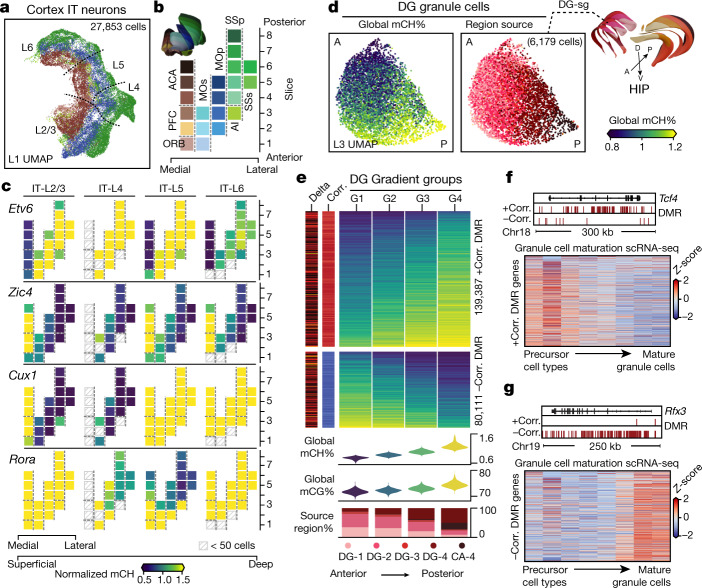
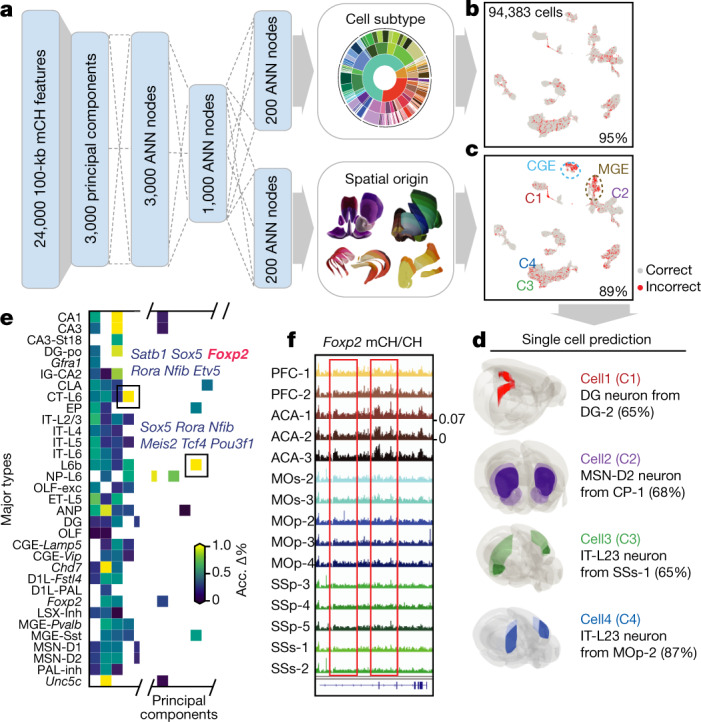
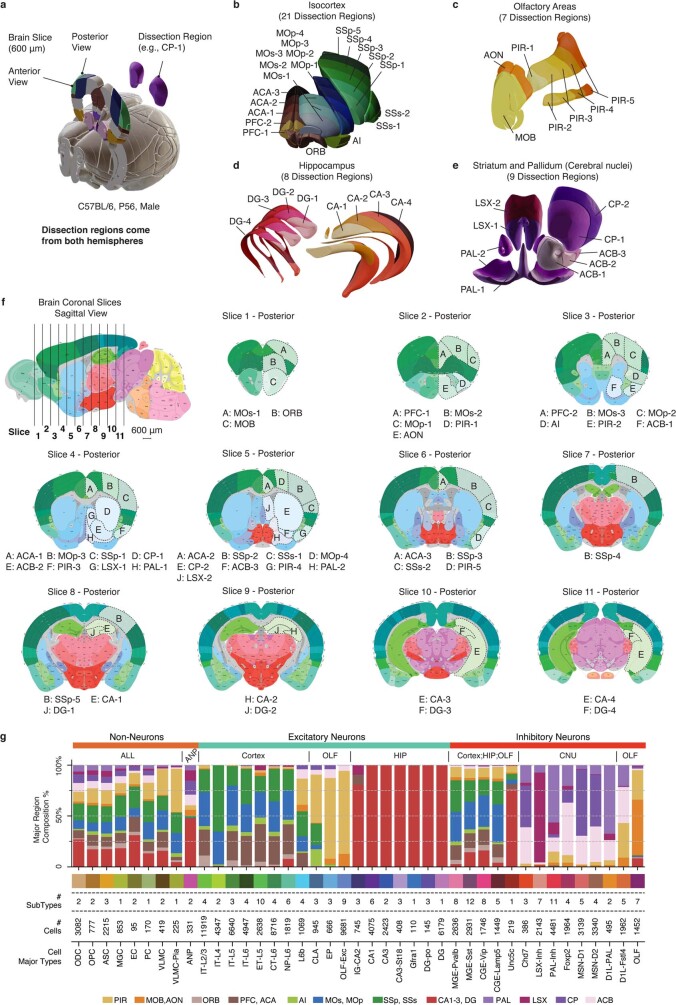
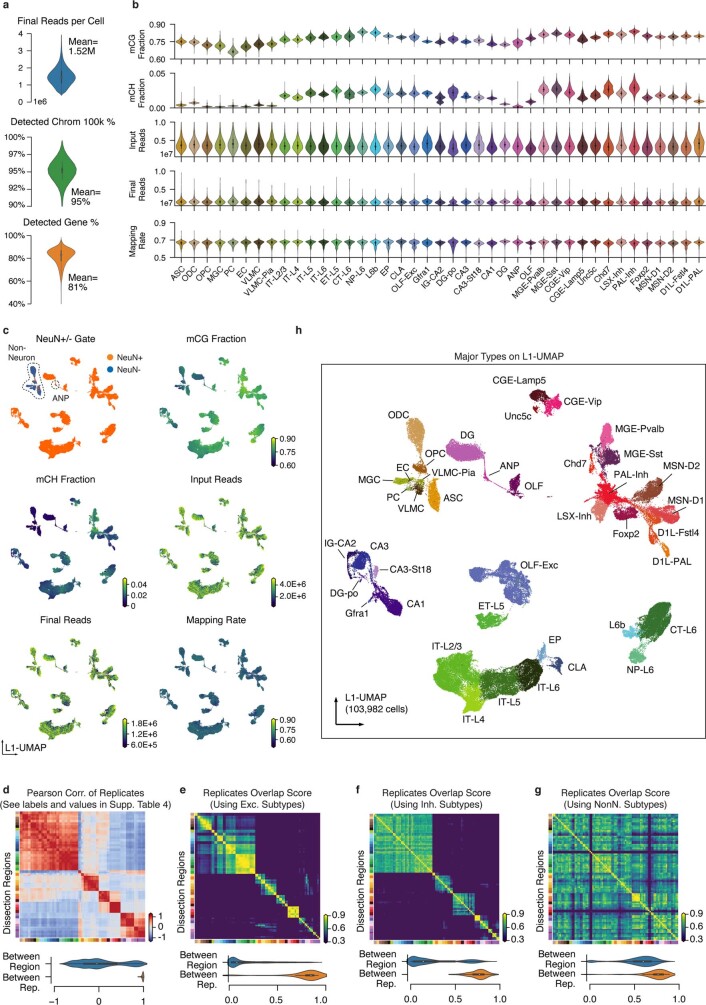
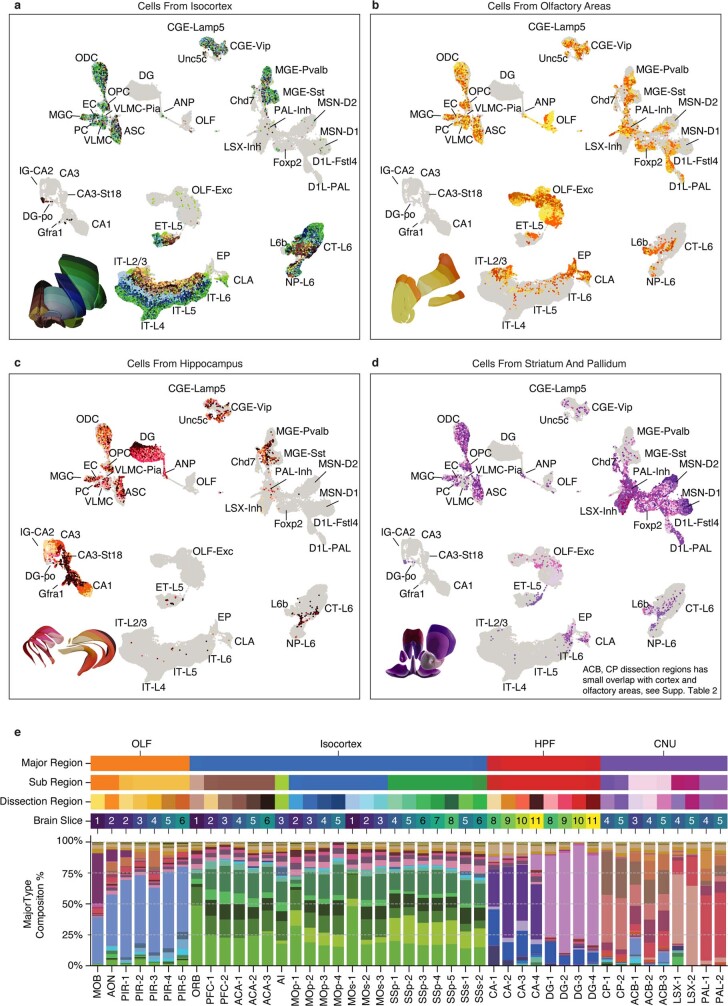
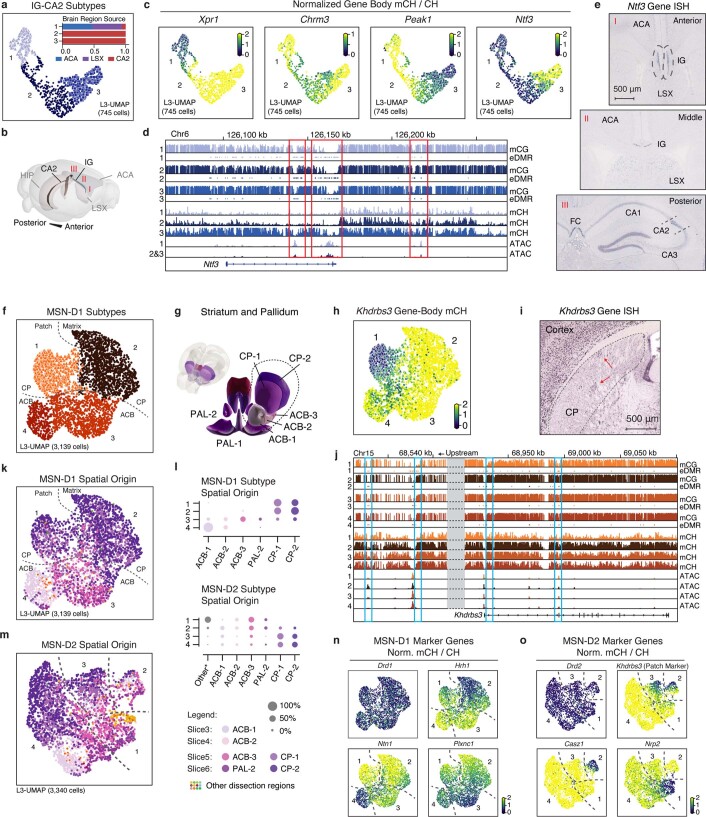
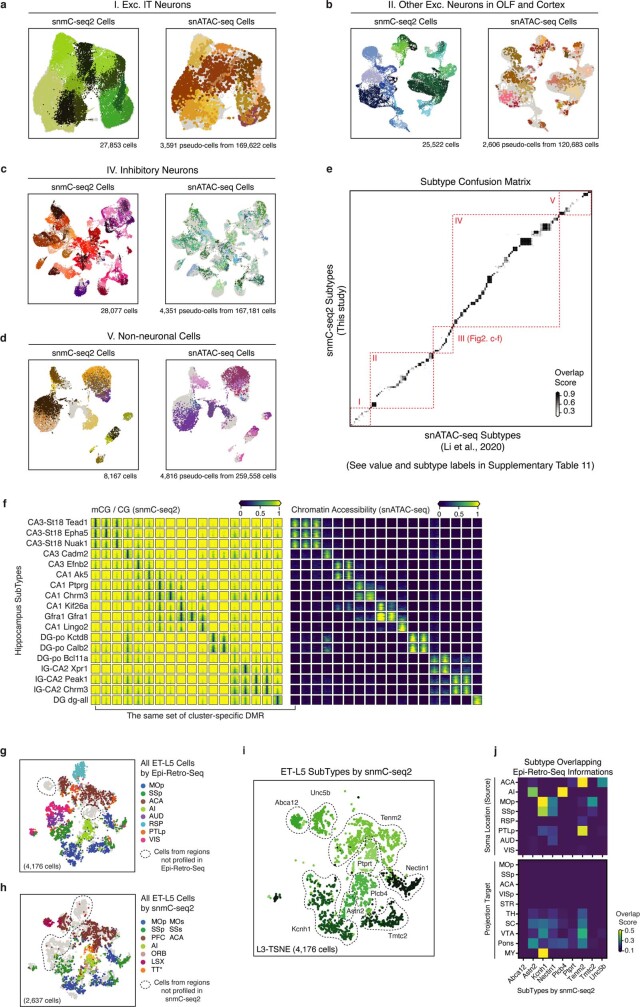
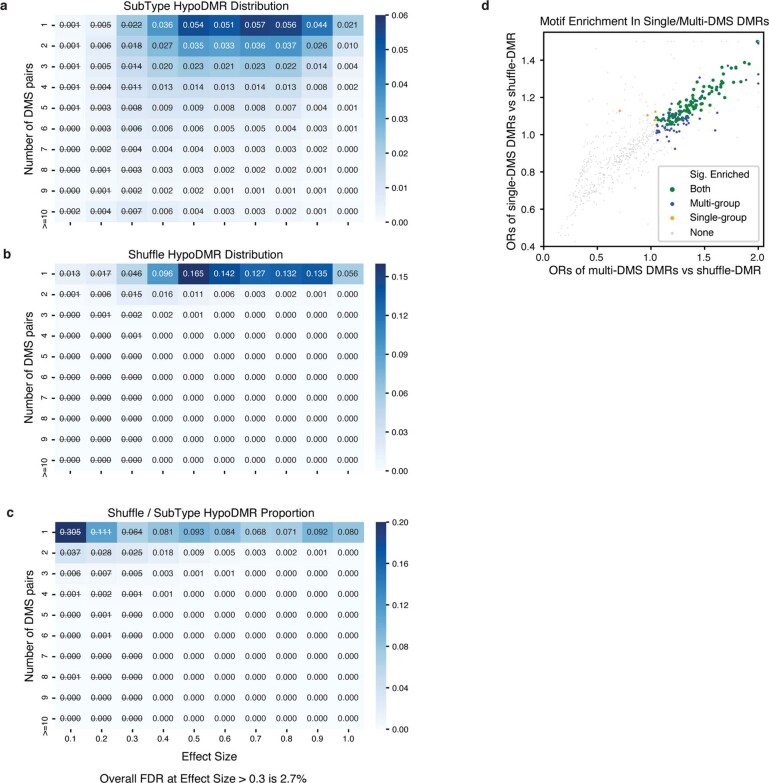
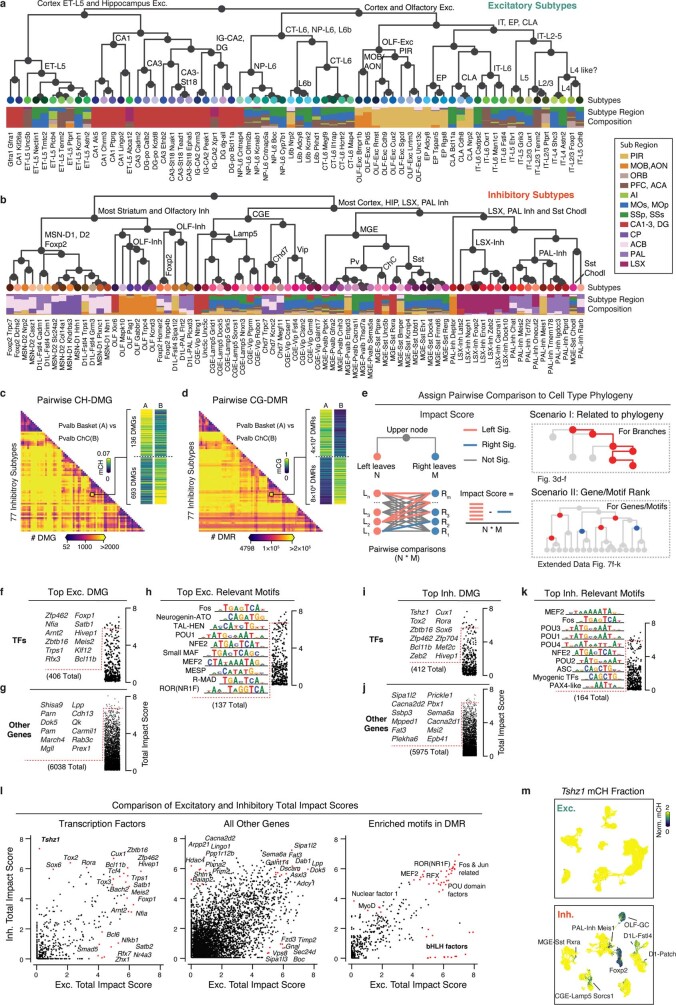
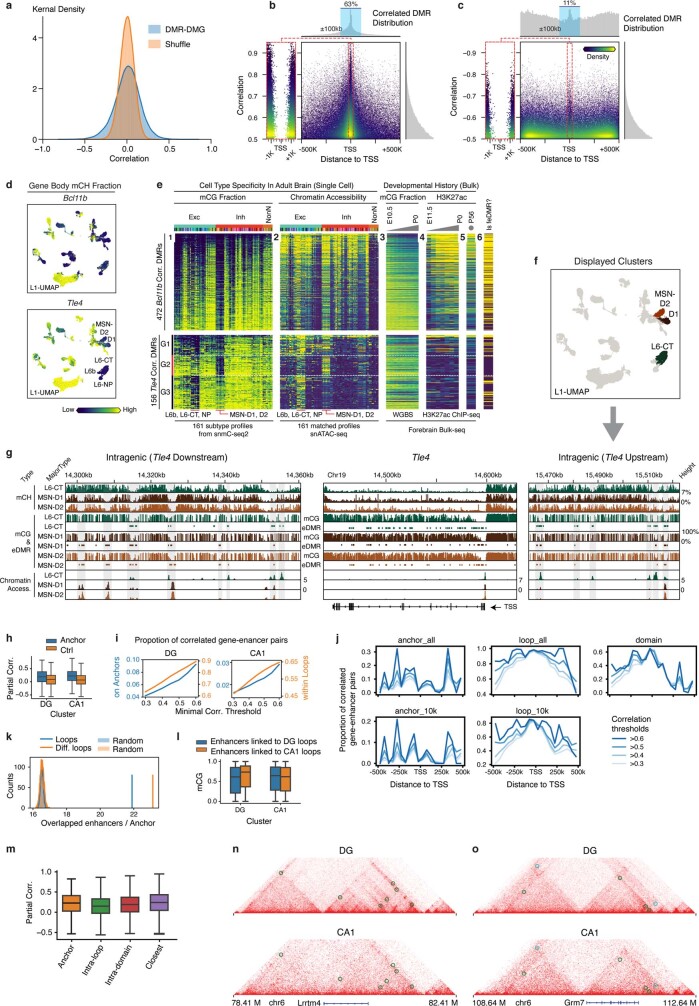
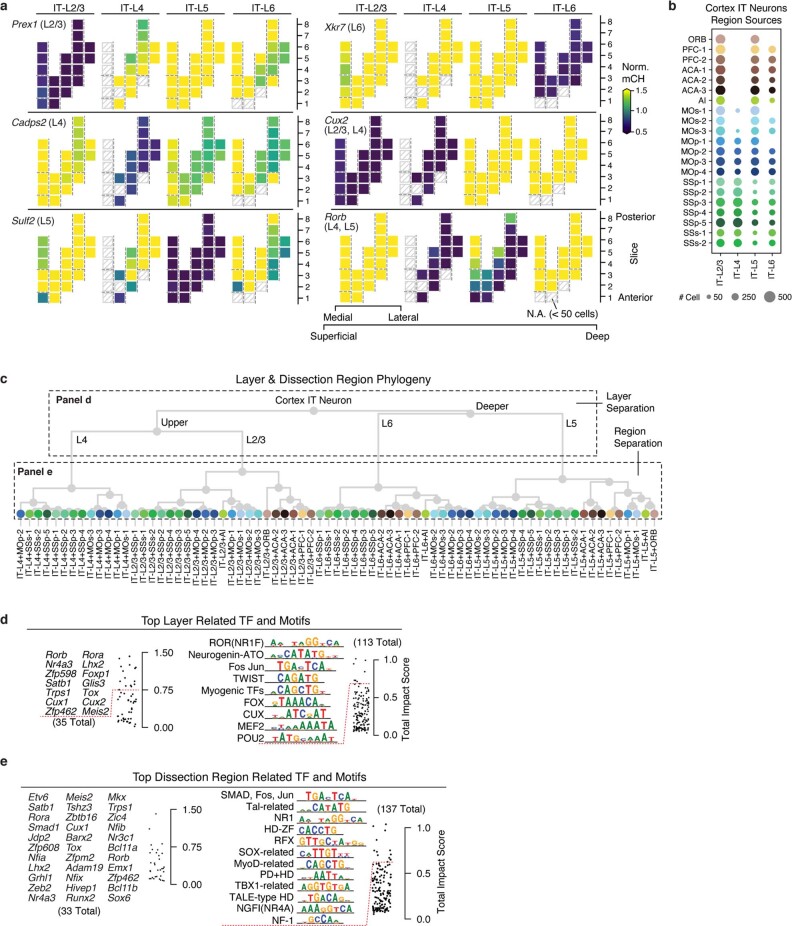
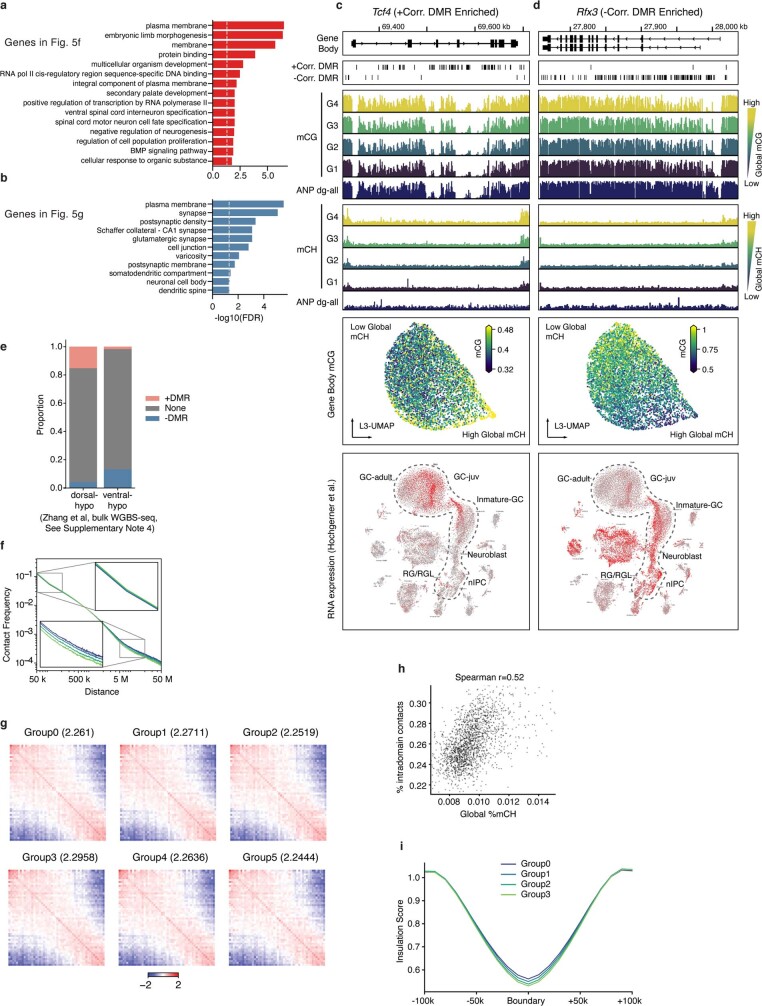
Mammalian brain cells show remarkable diversity in gene expression, anatomy and function, yet the regulatory DNA landscape underlying this extensive heterogeneity is poorly understood. Here we carry out a comprehensive assessment of the epigenomes of mouse brain cell types by applying single-nucleus DNA methylation sequencing1,2 to profile 103,982 nuclei (including 95,815 neurons and 8,167 non-neuronal cells) from 45 regions of the mouse cortex, hippocampus, striatum, pallidum and olfactory areas. We identified 161 cell clusters with distinct spatial locations and projection targets. We constructed taxonomies of these epigenetic types, annotated with signature genes, regulatory elements and transcription factors. These features indicate the potential regulatory landscape supporting the assignment of putative cell types and reveal repetitive usage of regulators in excitatory and inhibitory cells for determining subtypes. The DNA methylation landscape of excitatory neurons in the cortex and hippocampus varied continuously along spatial gradients. Using this deep dataset, we constructed an artificial neural network model that precisely predicts single neuron cell-type identity and brain area spatial location. Integration of high-resolution DNA methylomes with single-nucleus chromatin accessibility data3 enabled prediction of high-confidence enhancer-gene interactions for all identified cell types, which were subsequently validated by cell-type-specific chromatin conformation capture experiments4. By combining multi-omic datasets (DNA methylation, chromatin contacts, and open chromatin) from single nuclei and annotating the regulatory genome of hundreds of cell types in the mouse brain, our DNA methylation atlas establishes the epigenetic basis for neuronal diversity and spatial organization throughout the mouse cerebrum.
© 2021. The Author(s).
Conflict of interest statement
J.R.E serves on the scientific advisory board of Zymo Research Inc. B.R. is a shareholder of Arima Genomics.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
