

A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex
- PMID: 34616066
- PMCID: PMC8494649
- DOI: 10.1038/s41586-021-03500-8
A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex
Abstract
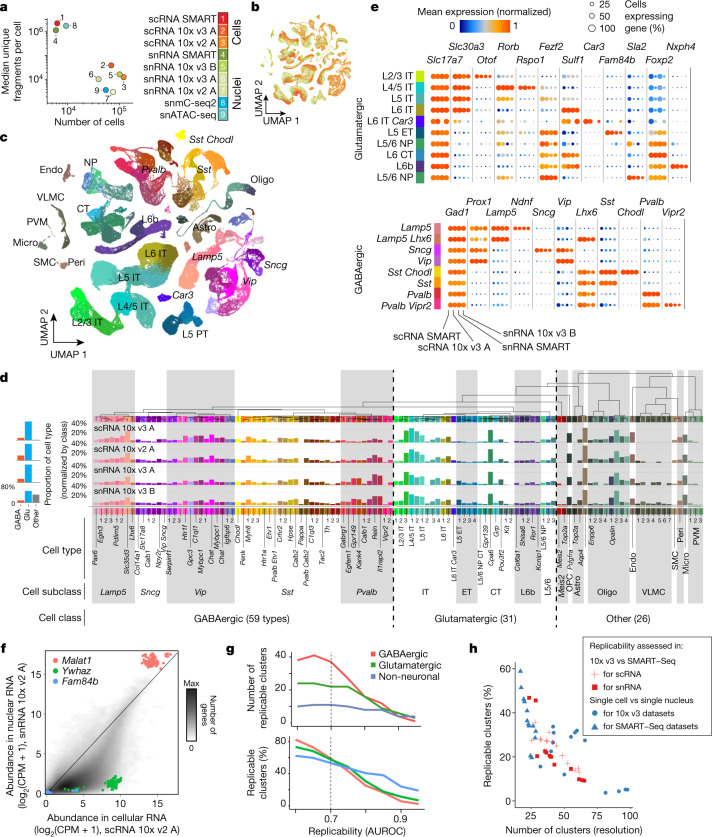
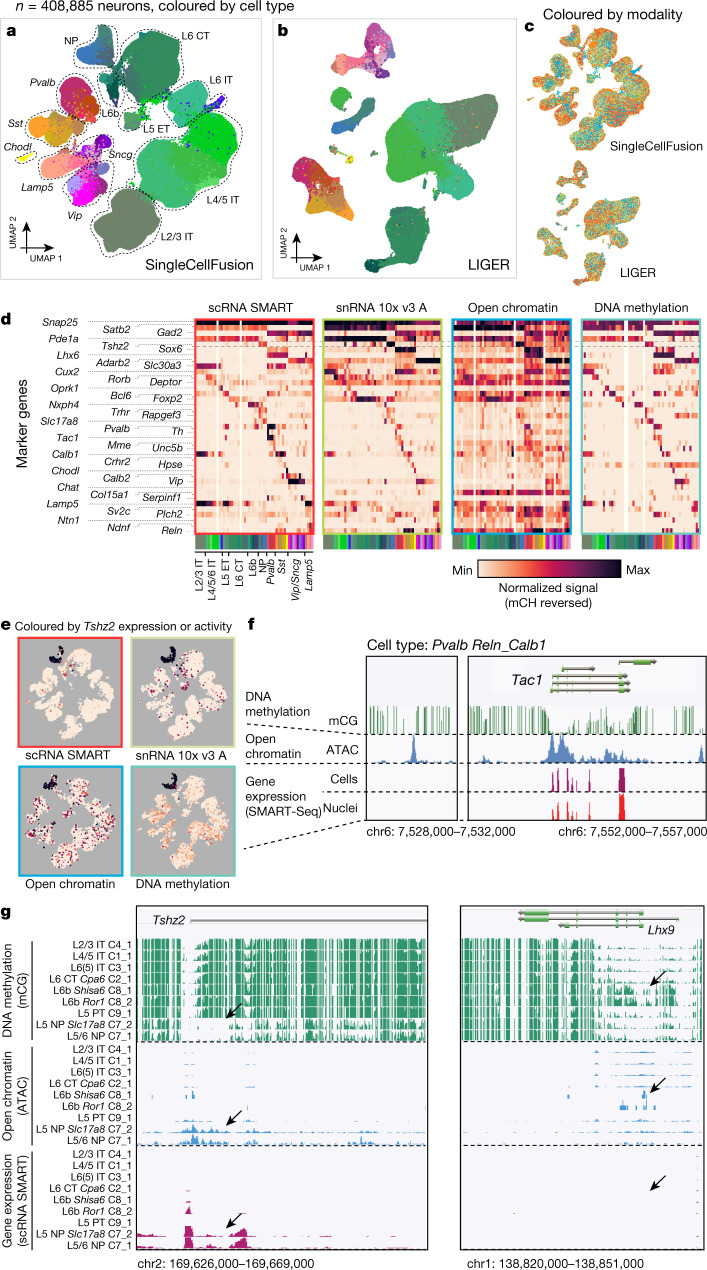
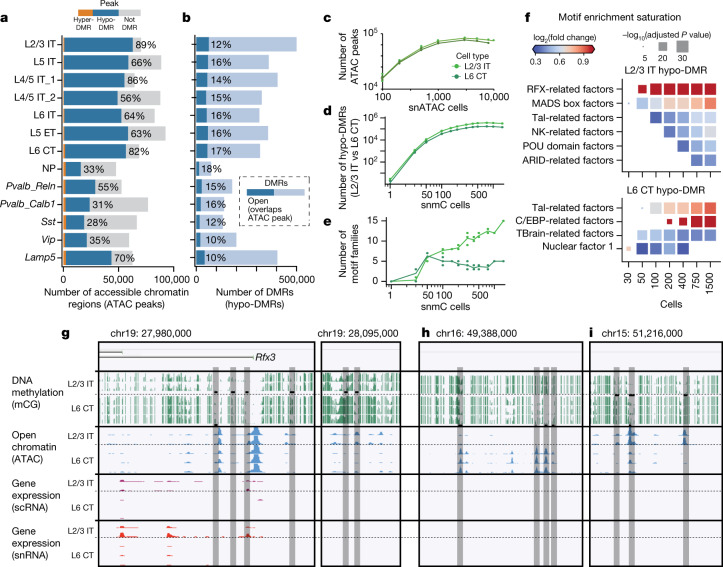
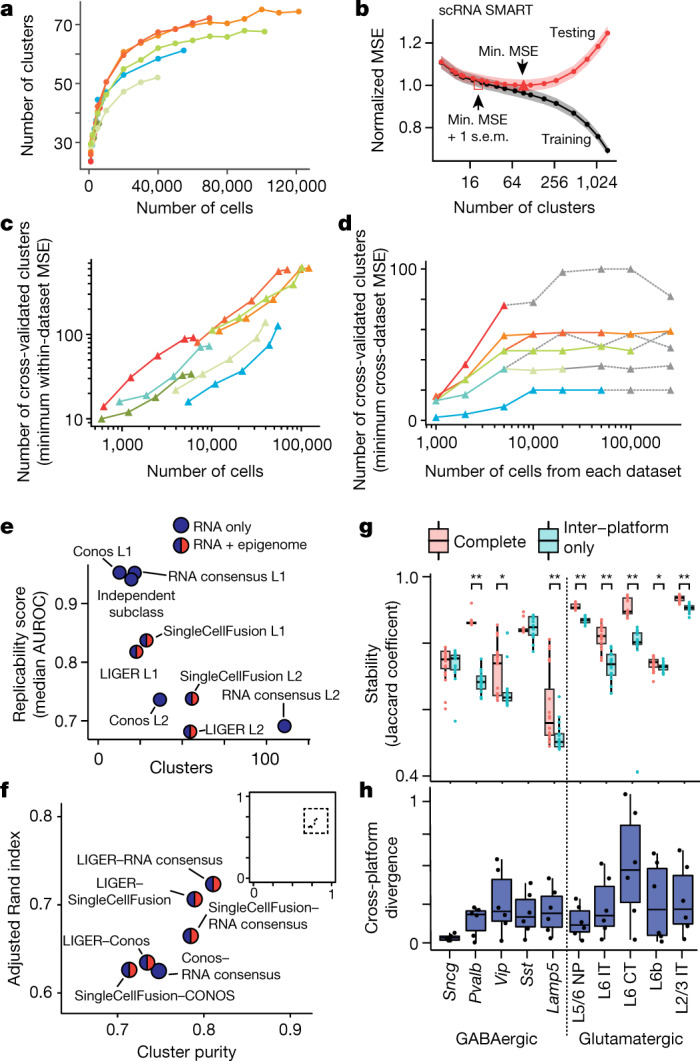
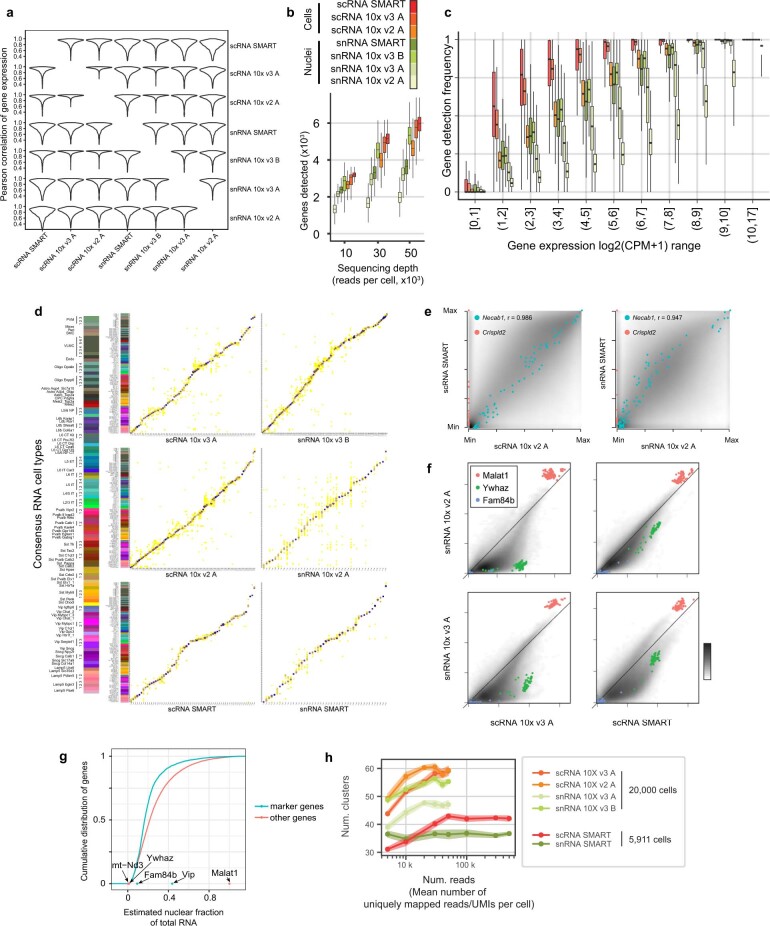
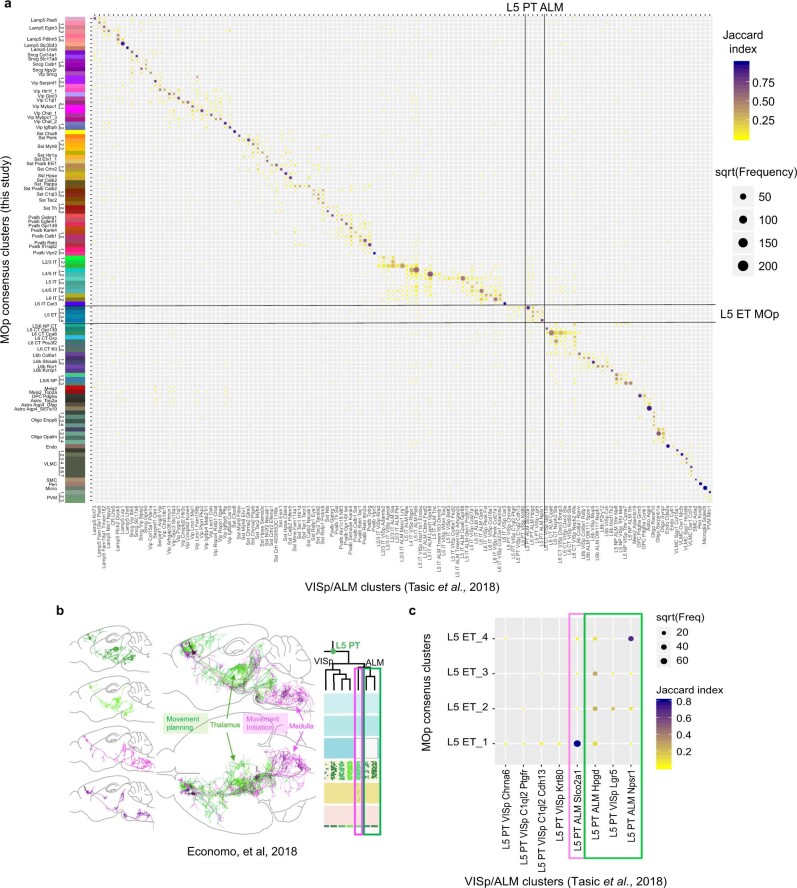
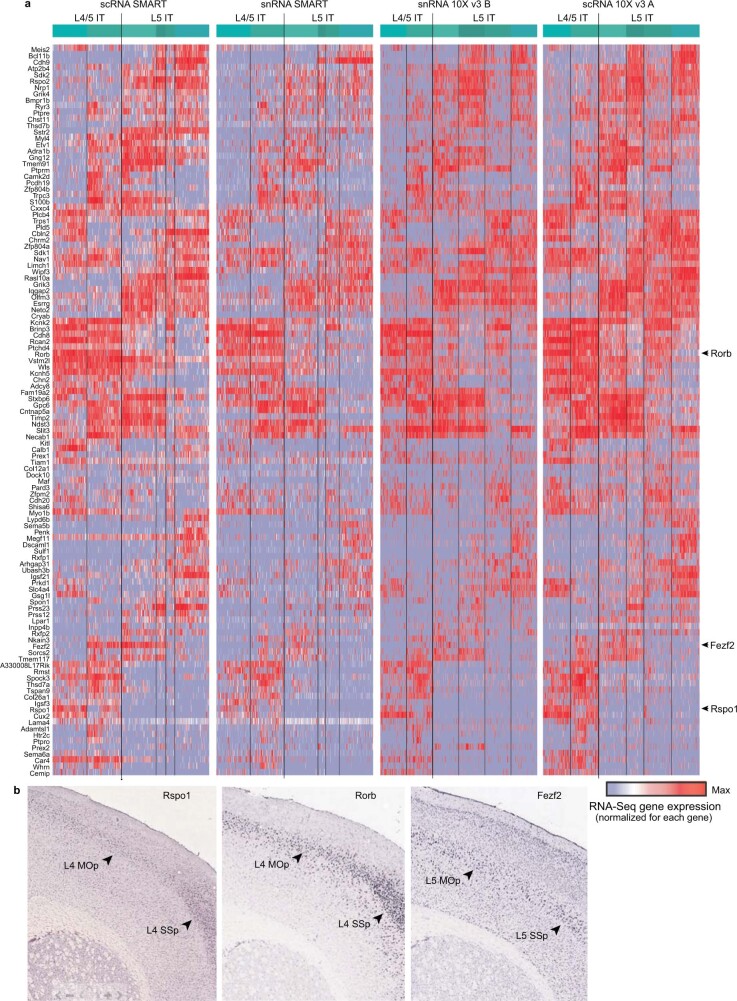
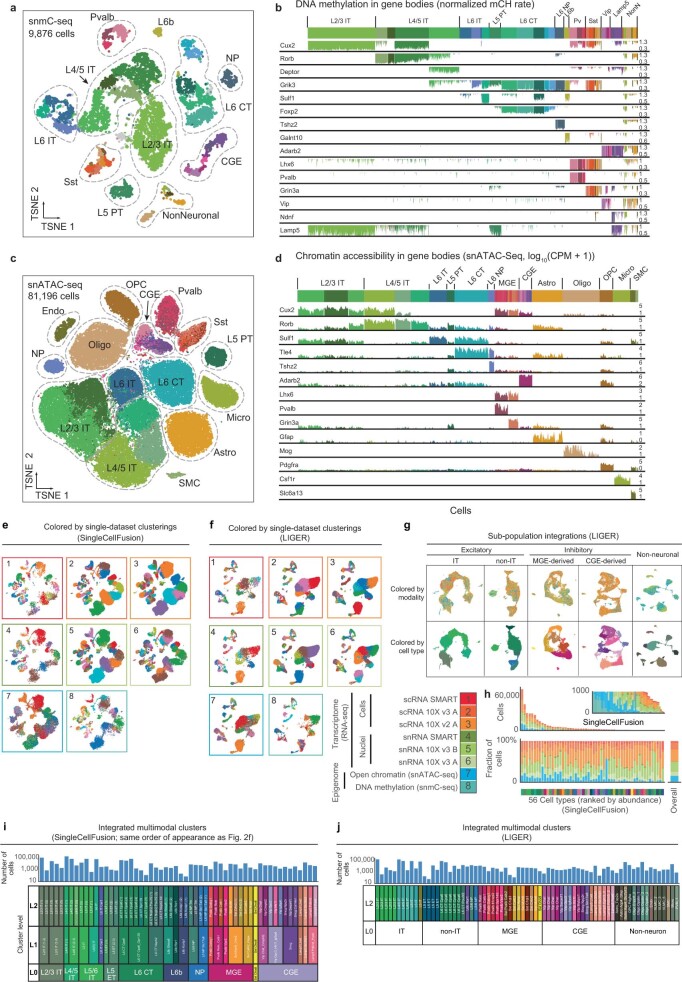
Single-cell transcriptomics can provide quantitative molecular signatures for large, unbiased samples of the diverse cell types in the brain1-3. With the proliferation of multi-omics datasets, a major challenge is to validate and integrate results into a biological understanding of cell-type organization. Here we generated transcriptomes and epigenomes from more than 500,000 individual cells in the mouse primary motor cortex, a structure that has an evolutionarily conserved role in locomotion. We developed computational and statistical methods to integrate multimodal data and quantitatively validate cell-type reproducibility. The resulting reference atlas-containing over 56 neuronal cell types that are highly replicable across analysis methods, sequencing technologies and modalities-is a comprehensive molecular and genomic account of the diverse neuronal and non-neuronal cell types in the mouse primary motor cortex. The atlas includes a population of excitatory neurons that resemble pyramidal cells in layer 4 in other cortical regions4. We further discovered thousands of concordant marker genes and gene regulatory elements for these cell types. Our results highlight the complex molecular regulation of cell types in the brain and will directly enable the design of reagents to target specific cell types in the mouse primary motor cortex for functional analysis.
© 2021. The Author(s).
Conflict of interest statement
B.R. is a shareholder of Arima Genomics, Inc. P.V.K. serves on the Scientific Advisory Board to Celsius Therapeutics, Inc. A.R. is an equity holder and founder of Celsius Therapeutics, an equity holder in Immunitas, and a Scientific Advisory Board member to Syros Pharmaceuticals, Neogene Therapeutics, Asimov and Thermo Fisher Scientific.
Figures


Comment in
-
A census of cell types in the brain's motor cortex.Nature. 2021 Oct;598(7879):33-34. doi: 10.1038/d41586-021-02493-8. Nature. 2021. PMID: 34616052 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- U19 MH114821/MH/NIMH NIH HHS/United States
- R24 MH114815/MH/NIMH NIH HHS/United States
- R01 DC013817/DC/NIDCD NIH HHS/United States
- U01 DC013817/DC/NIDCD NIH HHS/United States
- DC013817/NH/NIH HHS/United States
- GM114267/NH/NIH HHS/United States
- RF1 MH123199/MH/NIMH NIH HHS/United States
- U01 MH121282/MH/NIMH NIH HHS/United States
- R24 MH114788/MH/NIMH NIH HHS/United States
- P30 CA014195/CA/NCI NIH HHS/United States
- R01 DC019370/DC/NIDCD NIH HHS/United States
- U24 MH114827/MH/NIMH NIH HHS/United States
- R01 GM114267/GM/NIGMS NIH HHS/United States
- U19 MH114830/MH/NIMH NIH HHS/United States
