

An atlas of cortical arealization identifies dynamic molecular signatures
- PMID: 34616070
- PMCID: PMC8494648
- DOI: 10.1038/s41586-021-03910-8
An atlas of cortical arealization identifies dynamic molecular signatures
Abstract
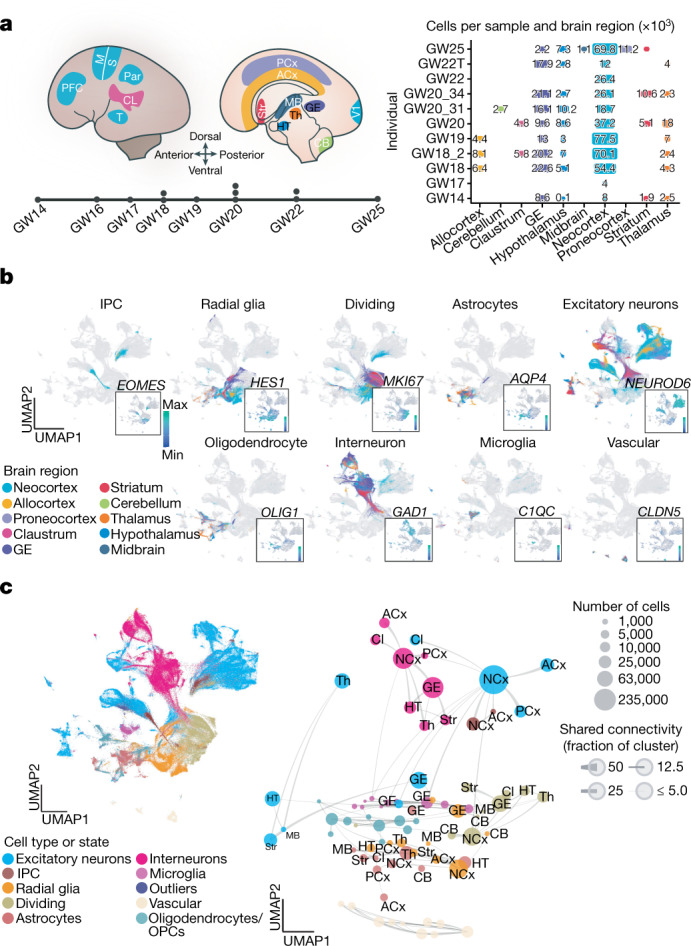
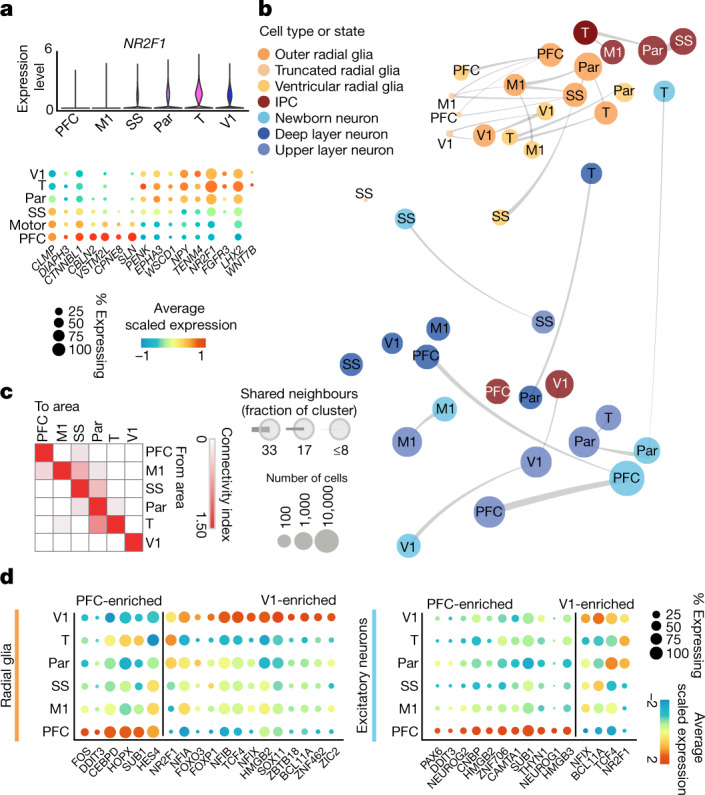
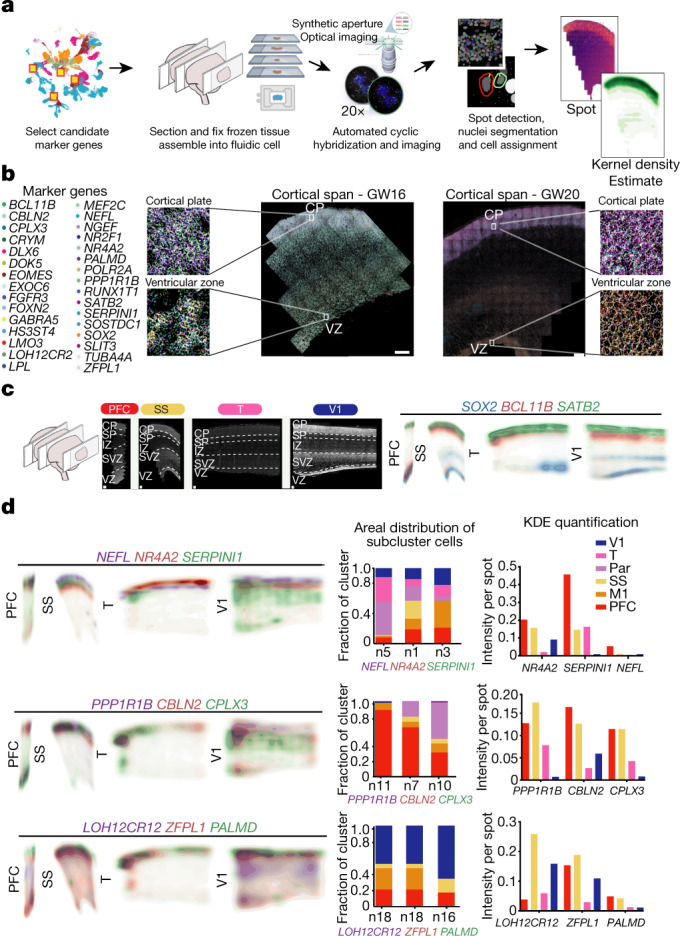
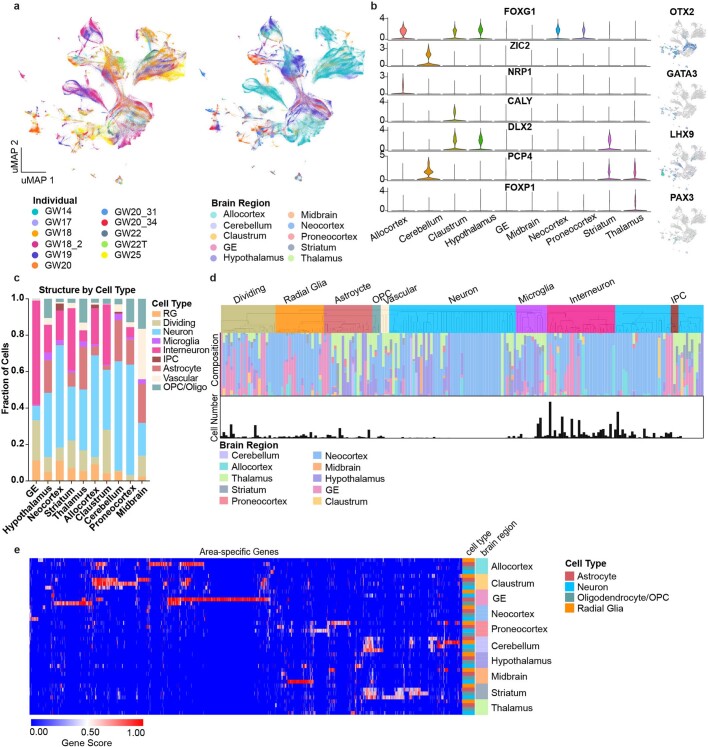
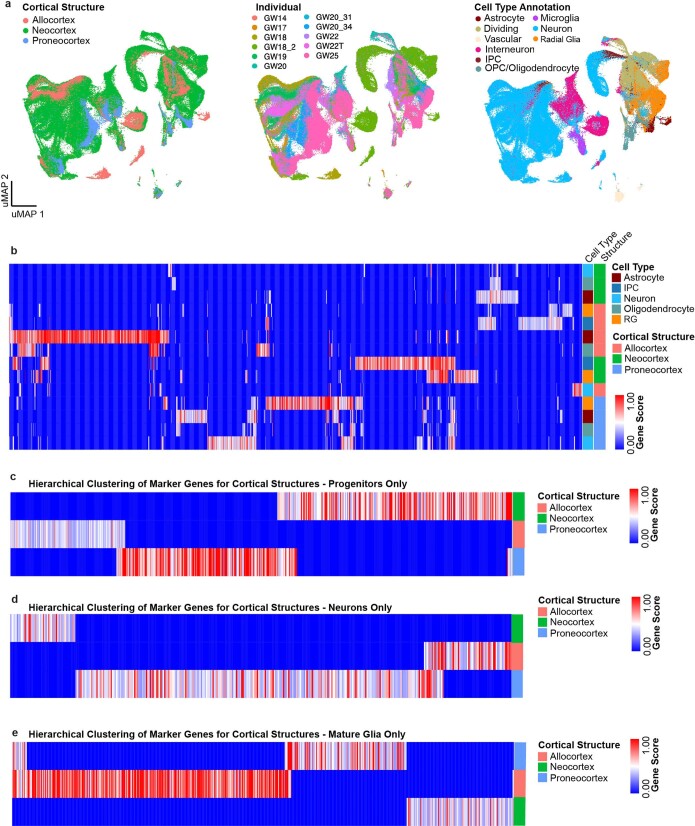
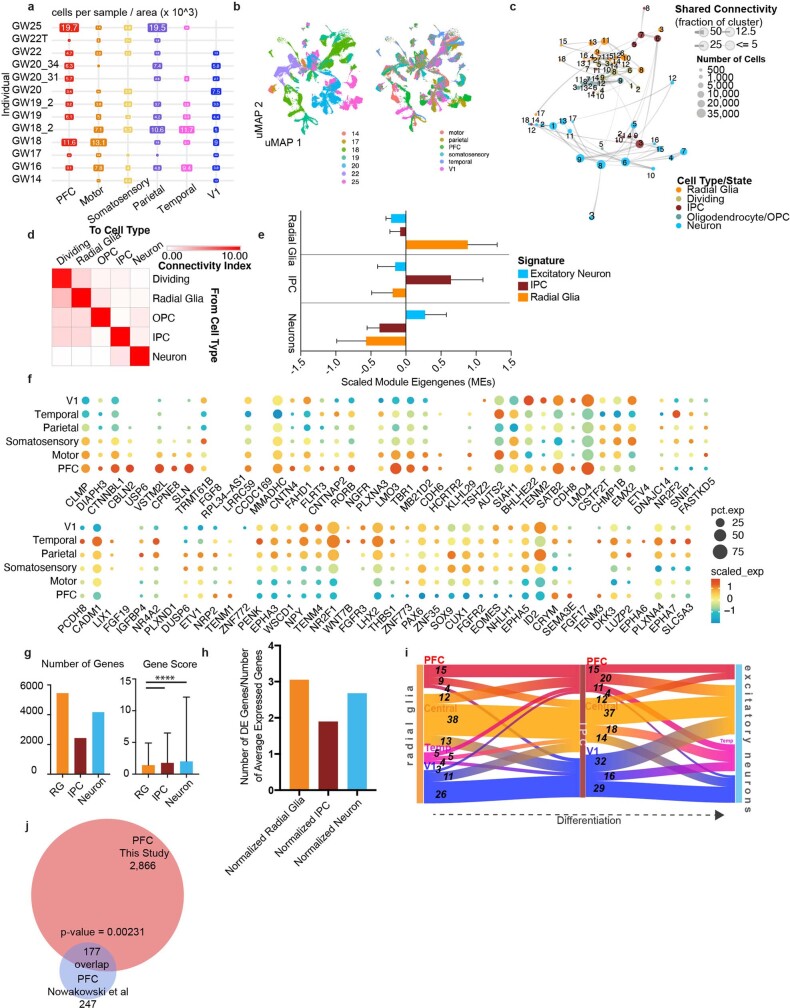
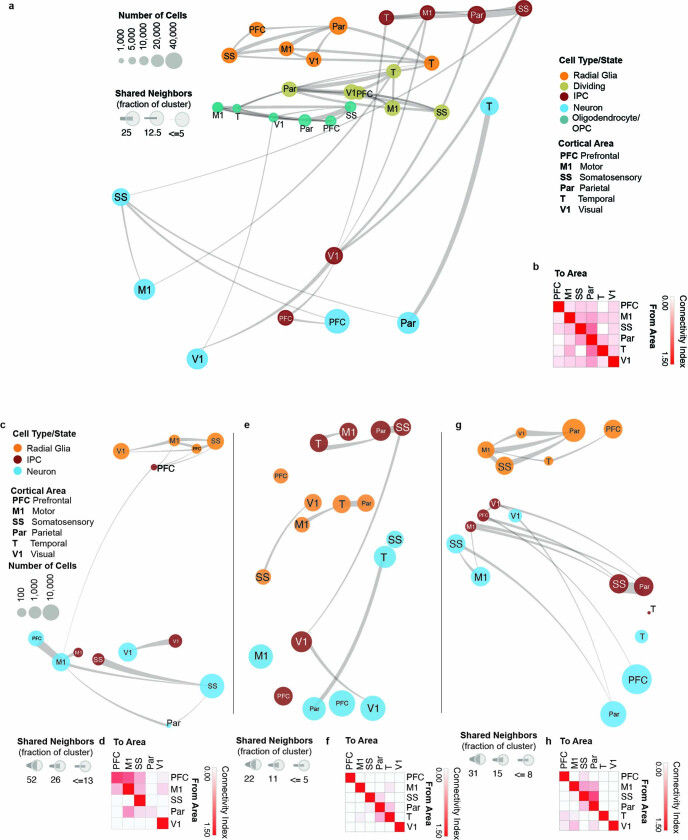
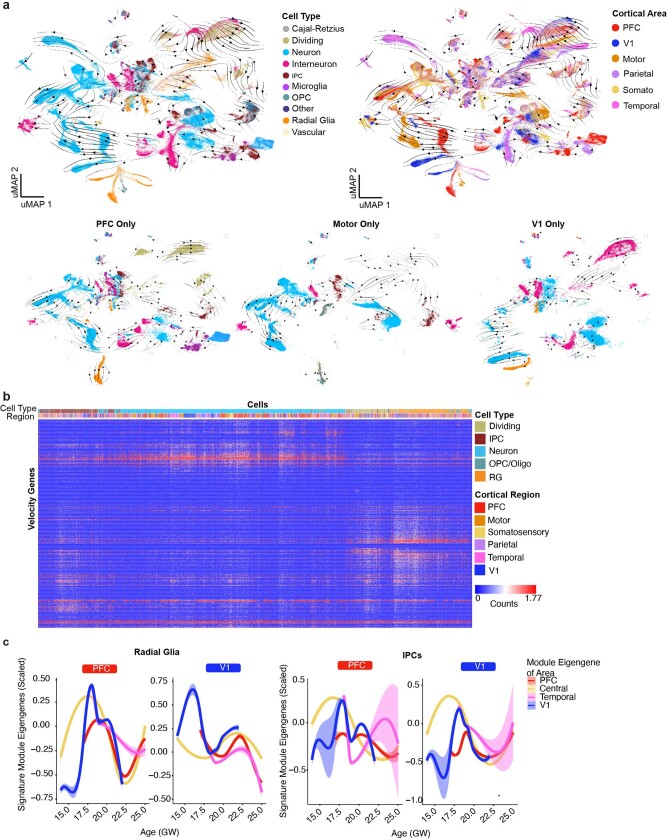
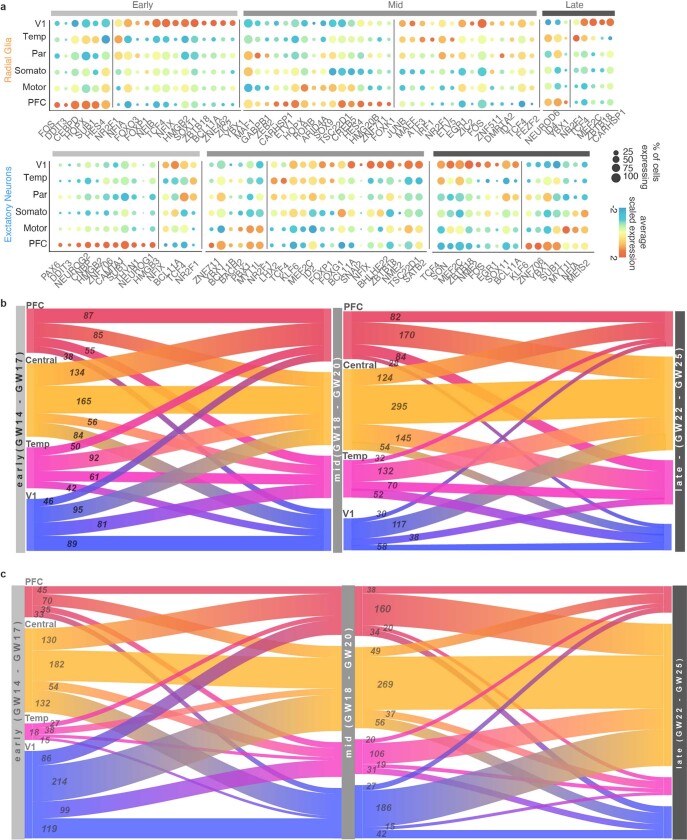
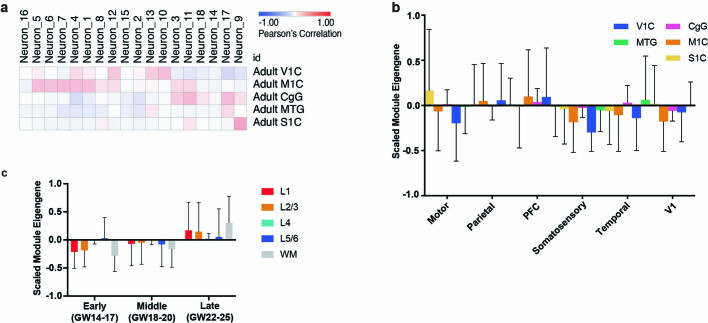
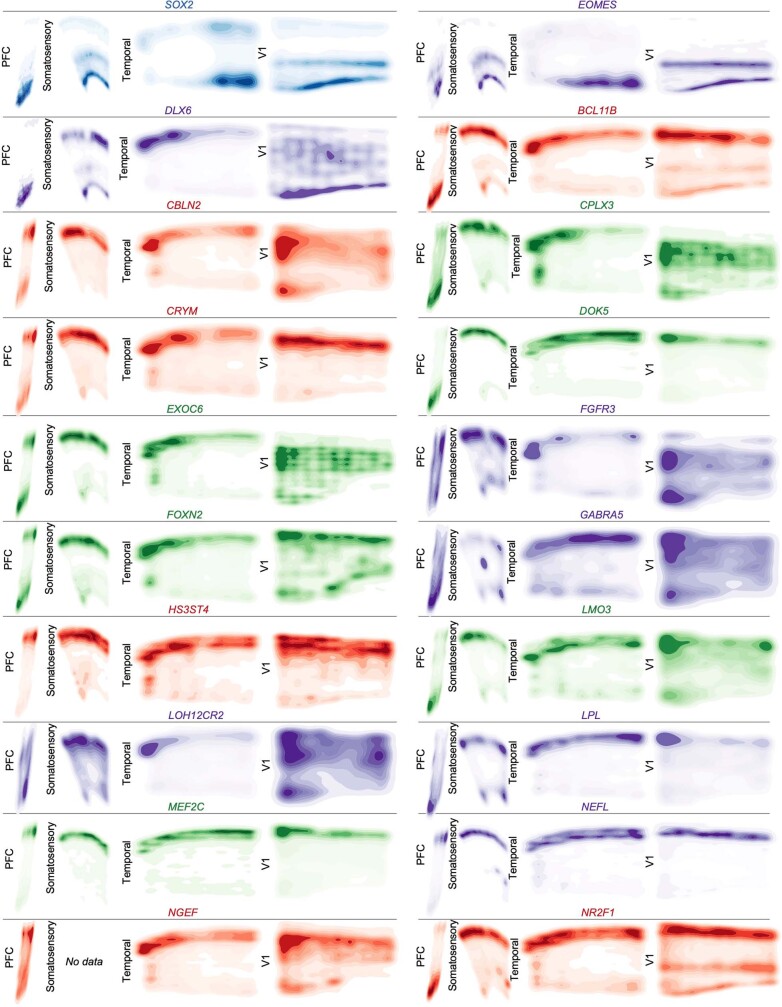
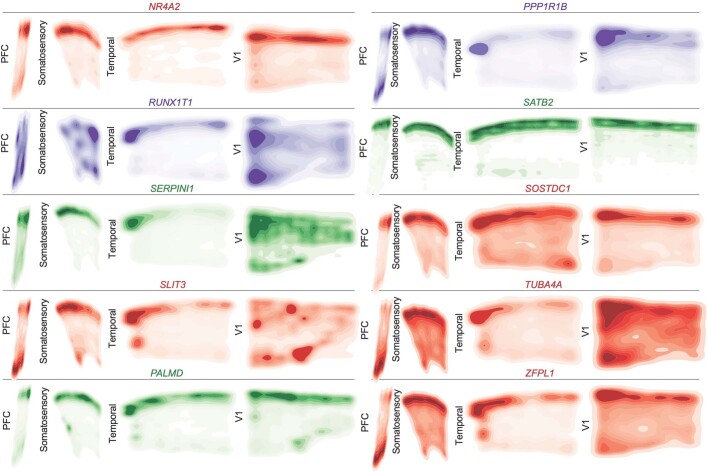
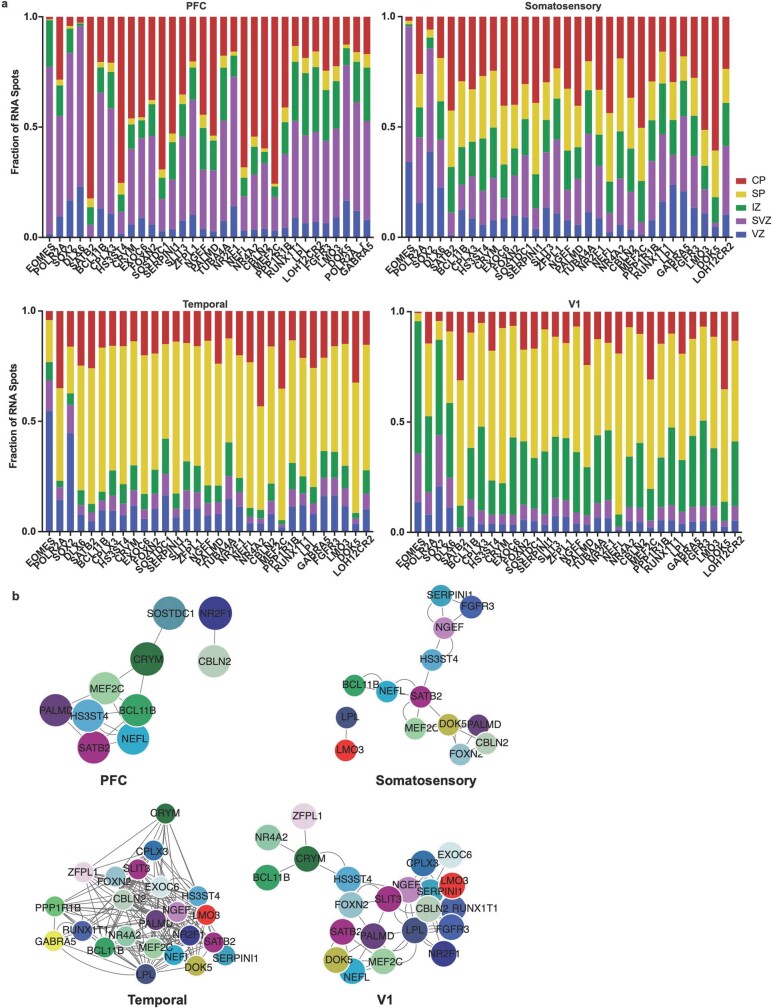
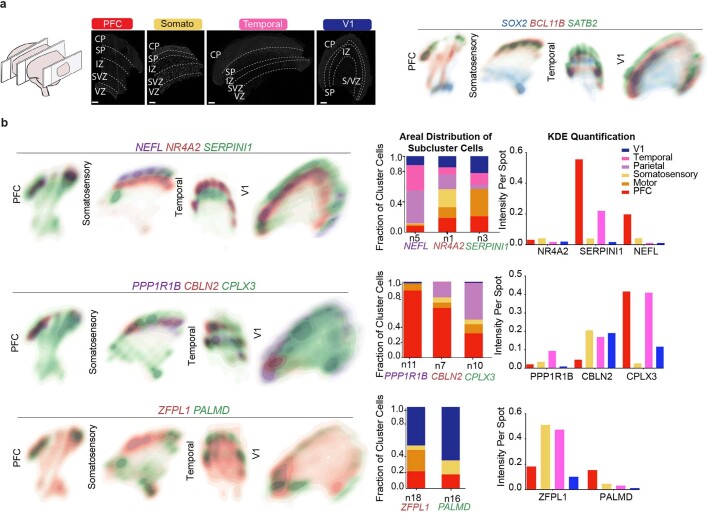
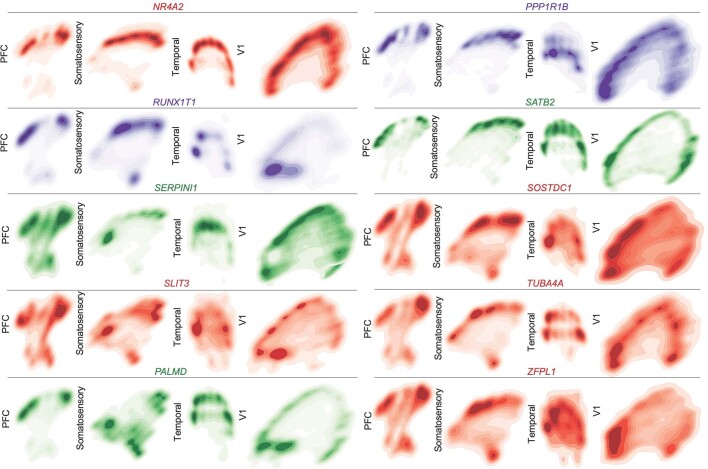
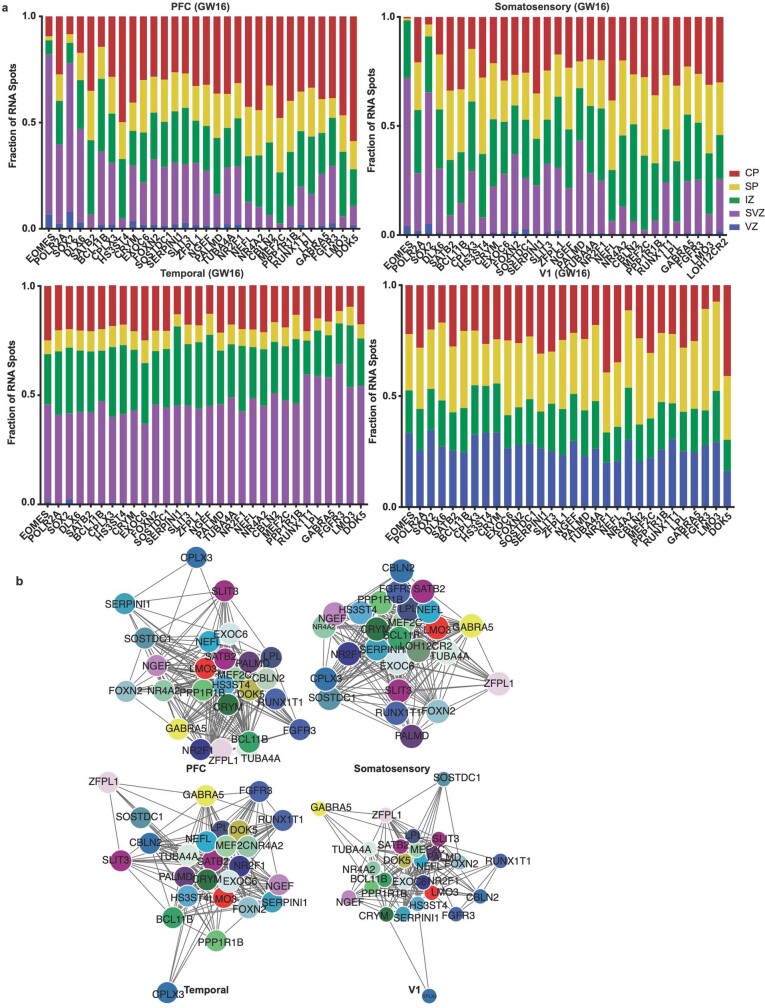
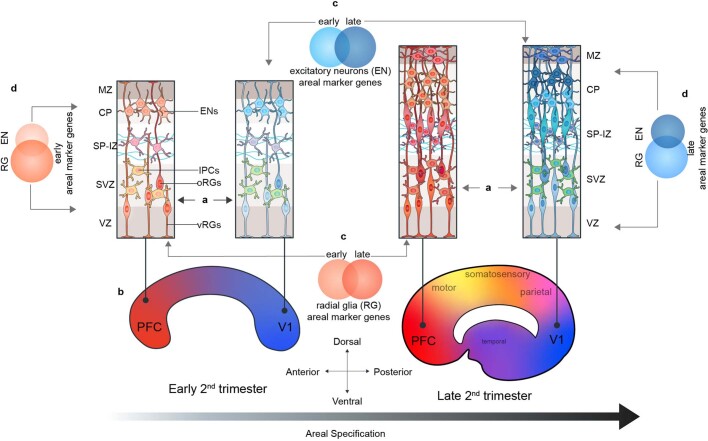
The human brain is subdivided into distinct anatomical structures, including the neocortex, which in turn encompasses dozens of distinct specialized cortical areas. Early morphogenetic gradients are known to establish early brain regions and cortical areas, but how early patterns result in finer and more discrete spatial differences remains poorly understood1. Here we use single-cell RNA sequencing to profile ten major brain structures and six neocortical areas during peak neurogenesis and early gliogenesis. Within the neocortex, we find that early in the second trimester, a large number of genes are differentially expressed across distinct cortical areas in all cell types, including radial glia, the neural progenitors of the cortex. However, the abundance of areal transcriptomic signatures increases as radial glia differentiate into intermediate progenitor cells and ultimately give rise to excitatory neurons. Using an automated, multiplexed single-molecule fluorescent in situ hybridization approach, we find that laminar gene-expression patterns are highly dynamic across cortical regions. Together, our data suggest that early cortical areal patterning is defined by strong, mutually exclusive frontal and occipital gene-expression signatures, with resulting gradients giving rise to the specification of areas between these two poles throughout successive developmental timepoints.
© 2021. The Author(s).
Conflict of interest statement
A.R.K. is a co-founder, consultant and director of Neurona Therapeutics. The remaining authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
