

Efficient and precise single-cell reference atlas mapping with Symphony
- PMID: 34620862
- PMCID: PMC8497570
- DOI: 10.1038/s41467-021-25957-x
Efficient and precise single-cell reference atlas mapping with Symphony
Abstract
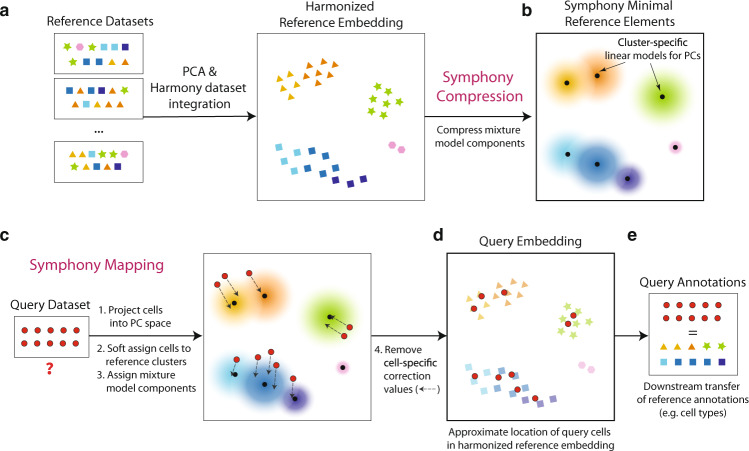
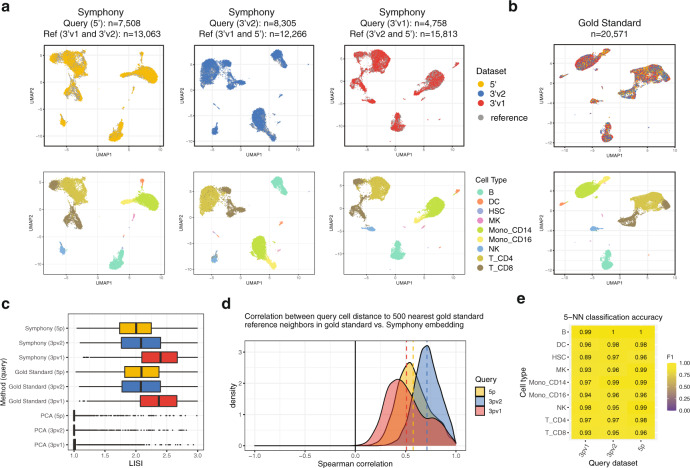
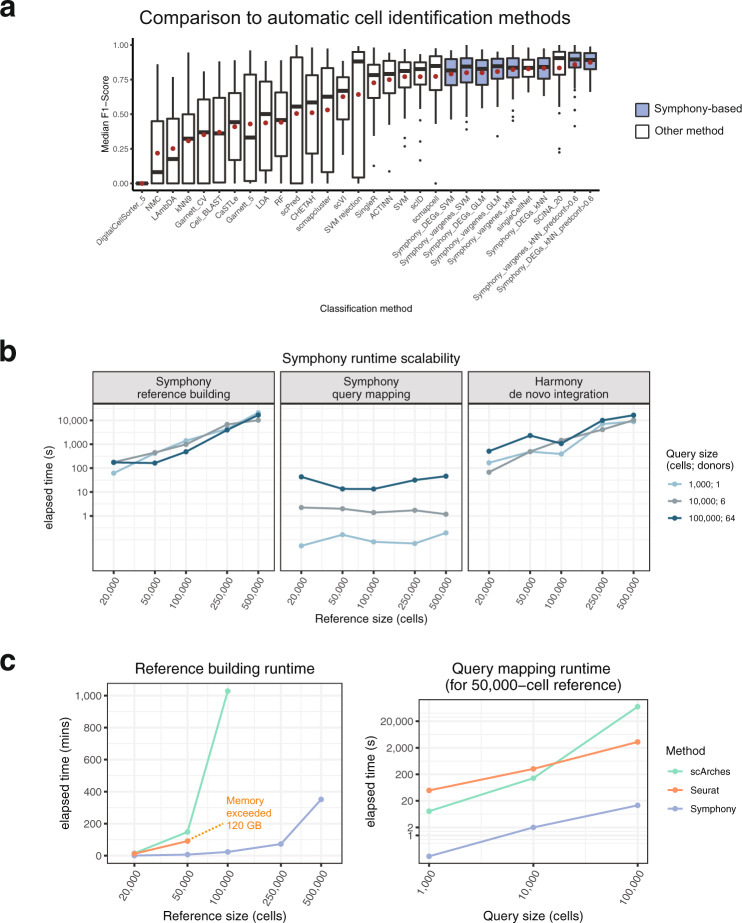
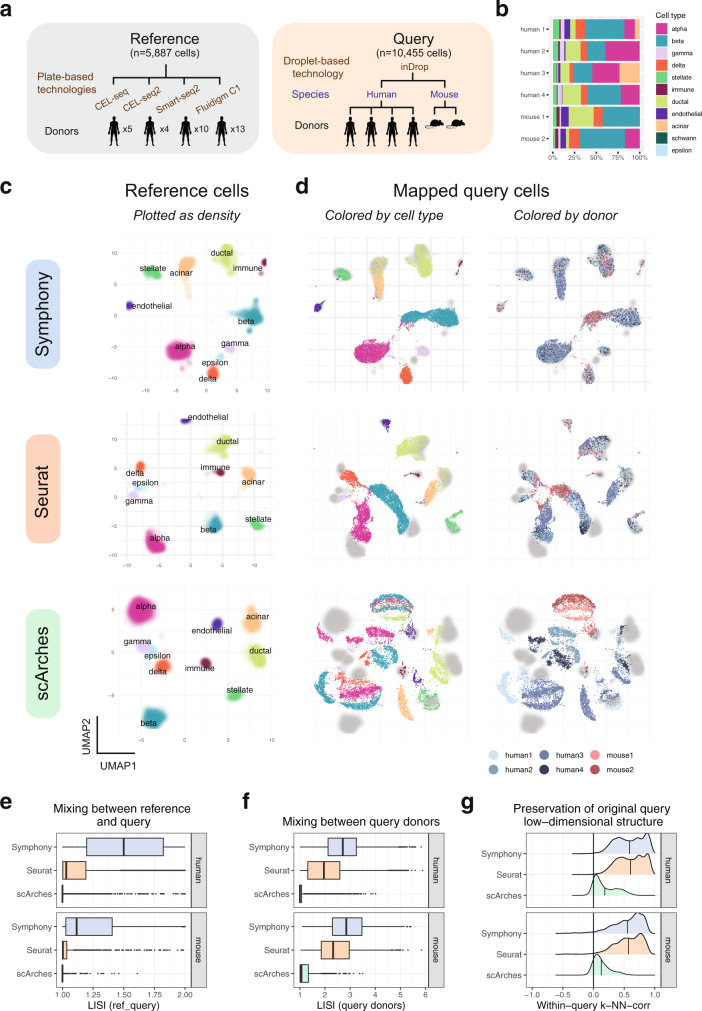
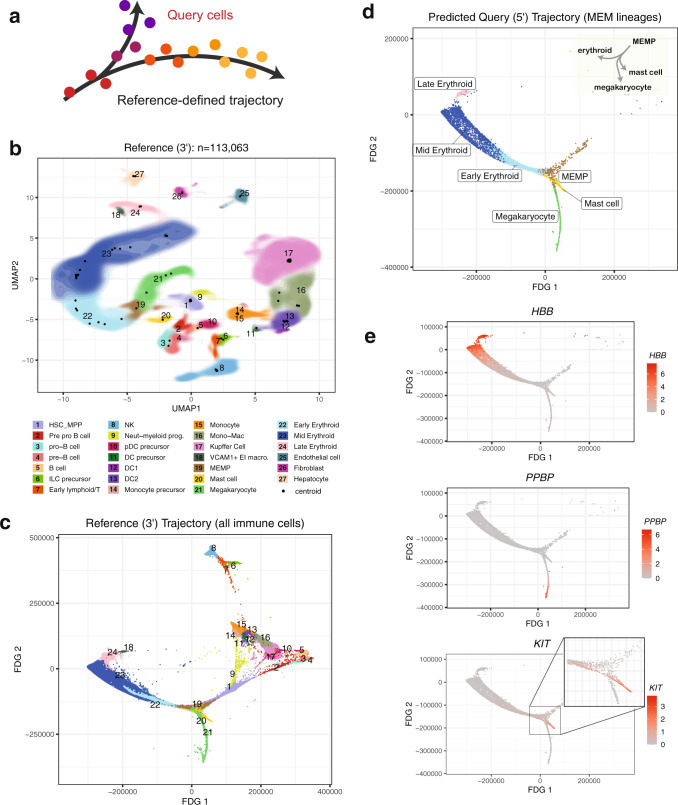
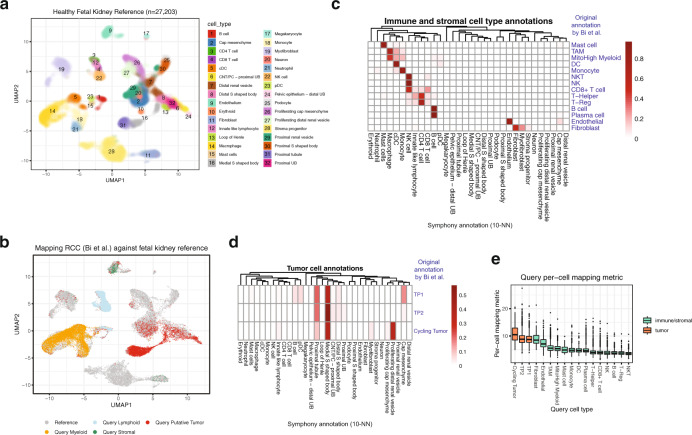
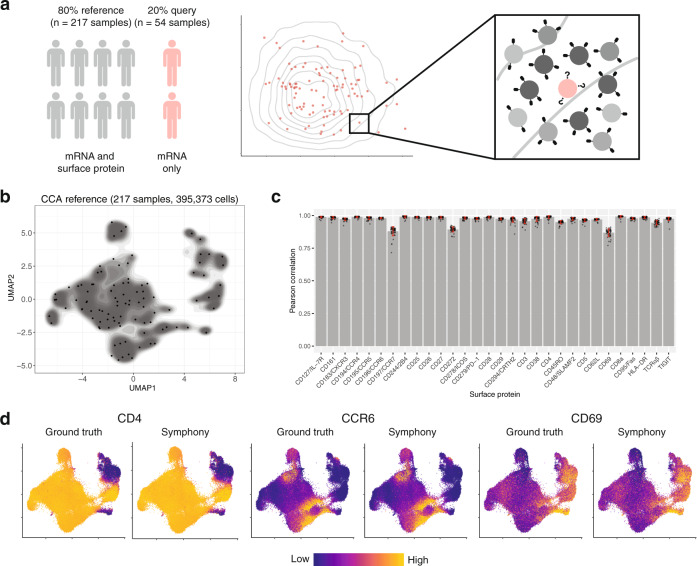
Recent advances in single-cell technologies and integration algorithms make it possible to construct comprehensive reference atlases encompassing many donors, studies, disease states, and sequencing platforms. Much like mapping sequencing reads to a reference genome, it is essential to be able to map query cells onto complex, multimillion-cell reference atlases to rapidly identify relevant cell states and phenotypes. We present Symphony ( https://github.com/immunogenomics/symphony ), an algorithm for building large-scale, integrated reference atlases in a convenient, portable format that enables efficient query mapping within seconds. Symphony localizes query cells within a stable low-dimensional reference embedding, facilitating reproducible downstream transfer of reference-defined annotations to the query. We demonstrate the power of Symphony in multiple real-world datasets, including (1) mapping a multi-donor, multi-species query to predict pancreatic cell types, (2) localizing query cells along a developmental trajectory of fetal liver hematopoiesis, and (3) inferring surface protein expression with a multimodal CITE-seq atlas of memory T cells.
© 2021. The Author(s).
Conflict of interest statement
S.R. receives research support from Biogen. I.K. does bioinformatics consulting for Brilyant Inc. No other authors have competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
