

Histiocytic disorders
- PMID: 34620874
- PMCID: PMC10031765
- DOI: 10.1038/s41572-021-00307-9
Histiocytic disorders
Abstract
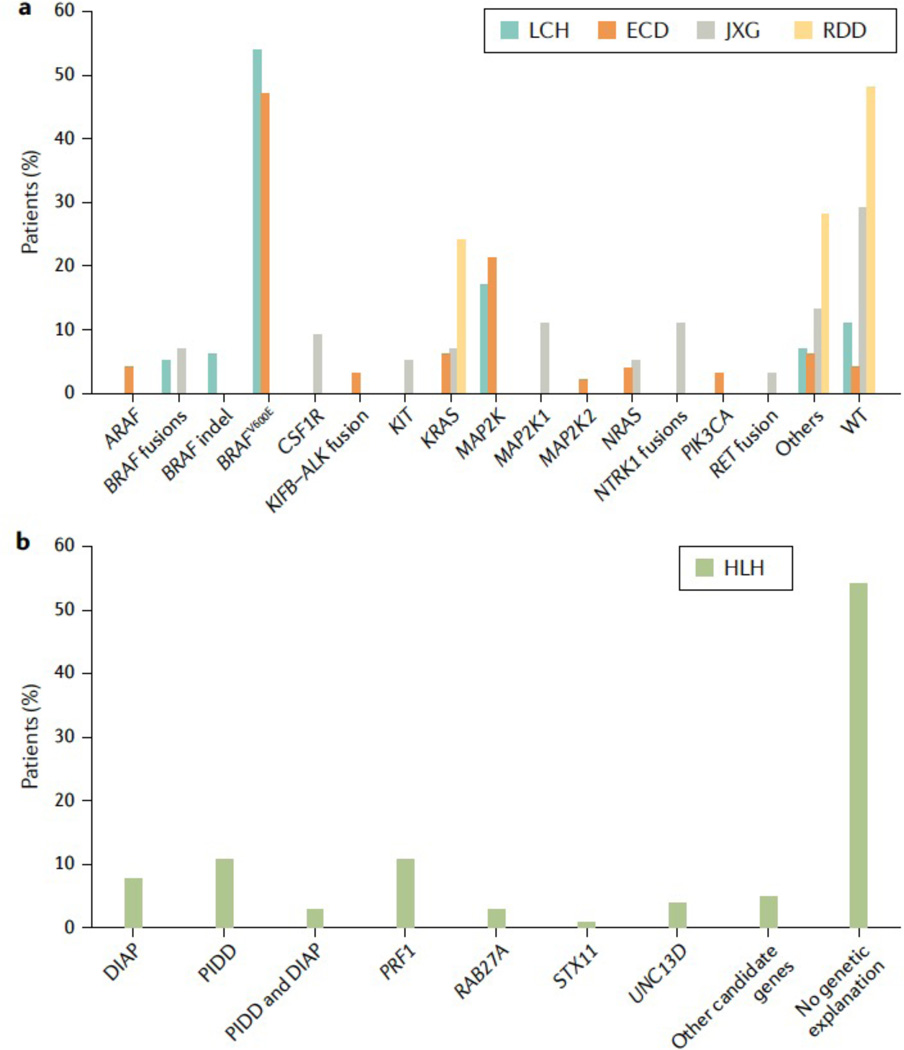
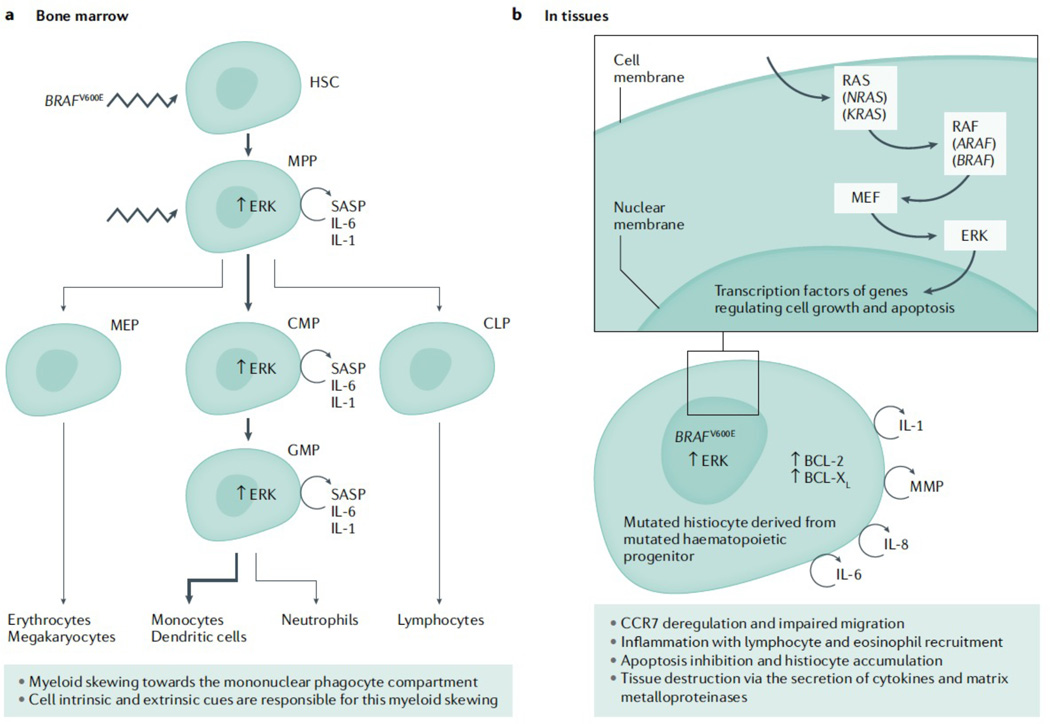
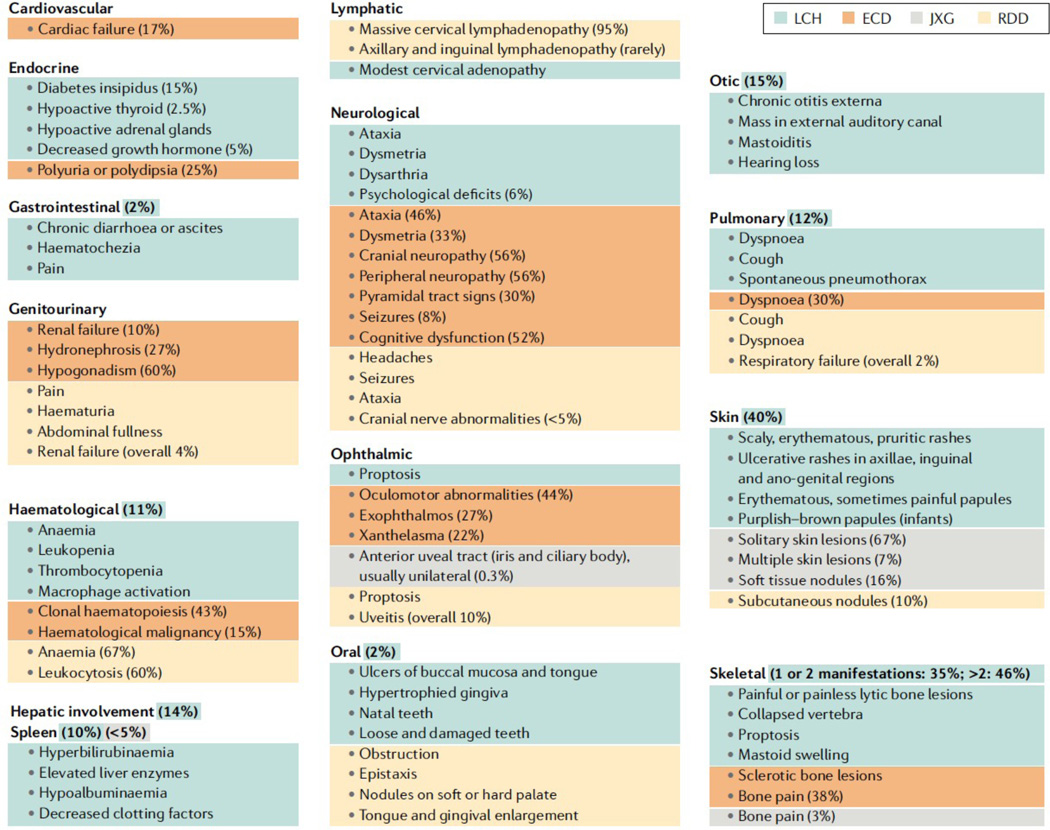
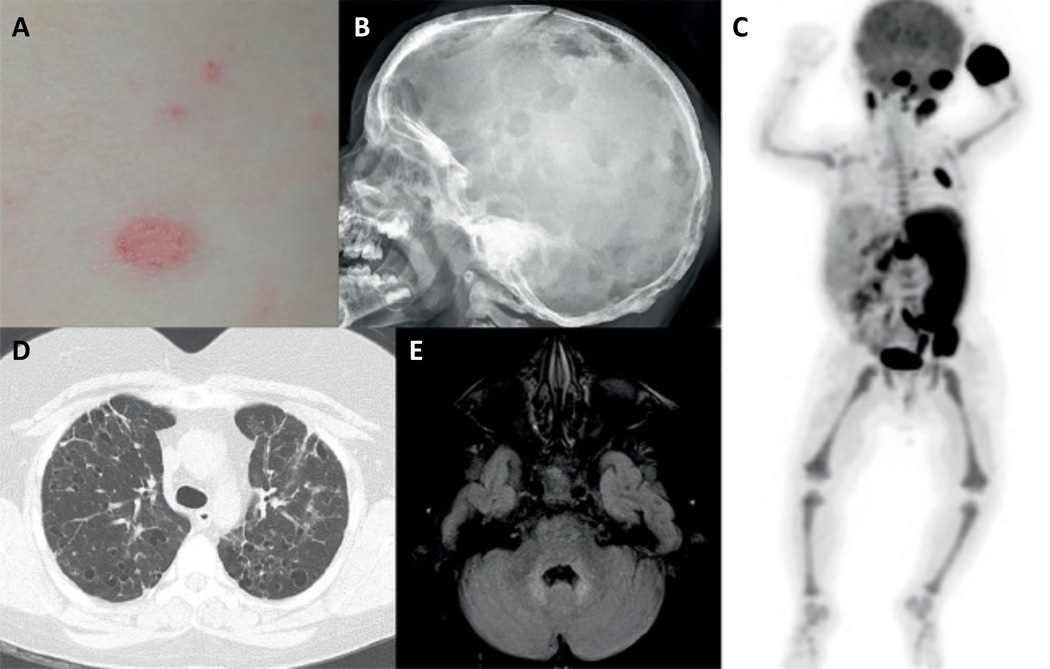
The historic term 'histiocytosis' meaning 'tissue cell' is used as a unifying concept for diseases characterized by pathogenic myeloid cells that share histological features with macrophages or dendritic cells. These cells may arise from the embryonic yolk sac, fetal liver or postnatal bone marrow. Prior classification schemes align disease designation with terminal phenotype: for example, Langerhans cell histiocytosis (LCH) shares CD207+ antigen with physiological epidermal Langerhans cells. LCH, Erdheim-Chester disease (ECD), juvenile xanthogranuloma (JXG) and Rosai-Dorfman disease (RDD) are all characterized by pathological ERK activation driven by activating somatic mutations in MAPK pathway genes. The title of this Primer (Histiocytic disorders) was chosen to differentiate the above diseases from Langerhans cell sarcoma and malignant histiocytosis, which are hyperproliferative lesions typical of cancer. By comparison LCH, ECD, RDD and JXG share some features of malignant cells including activating MAPK pathway mutations, but are not hyperproliferative. 'Inflammatory myeloproliferative neoplasm' may be a more precise nomenclature. By contrast, haemophagocytic lymphohistiocytosis is associated with macrophage activation and extreme inflammation, and represents a syndrome of immune dysregulation. These diseases affect children and adults in varying proportions depending on which of the entities is involved.
© 2021. Springer Nature Limited.
Figures
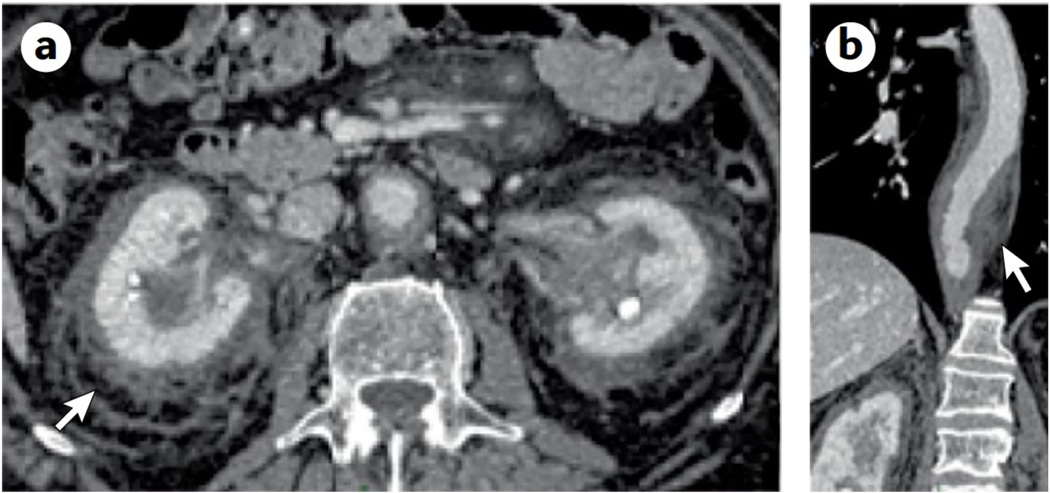
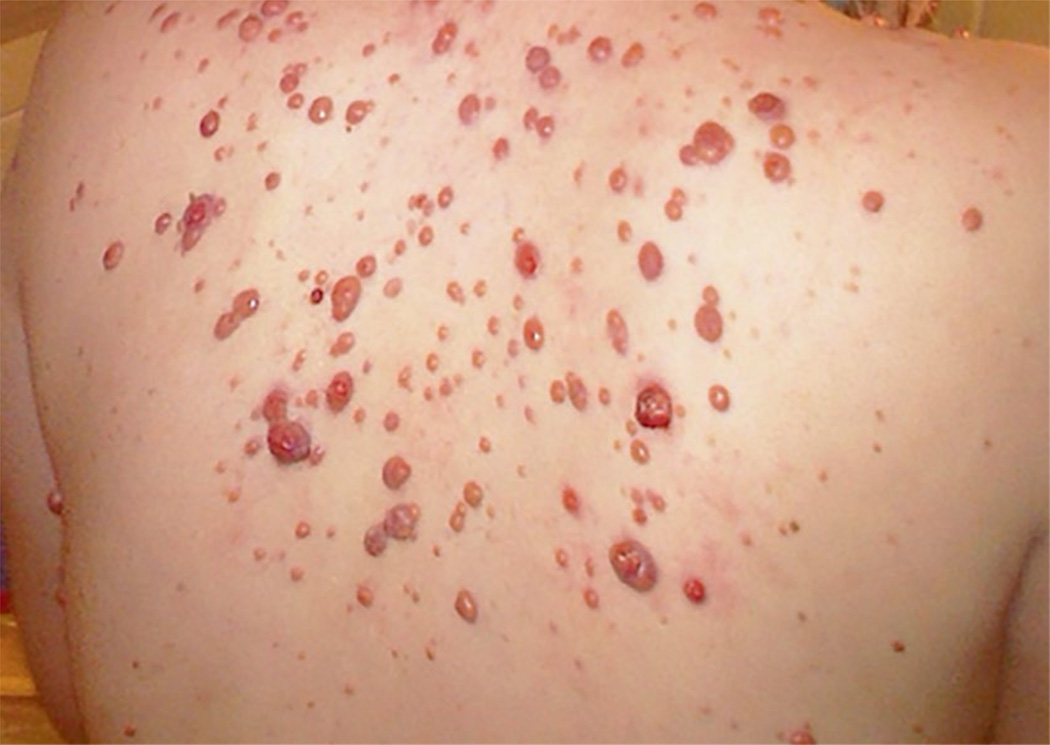
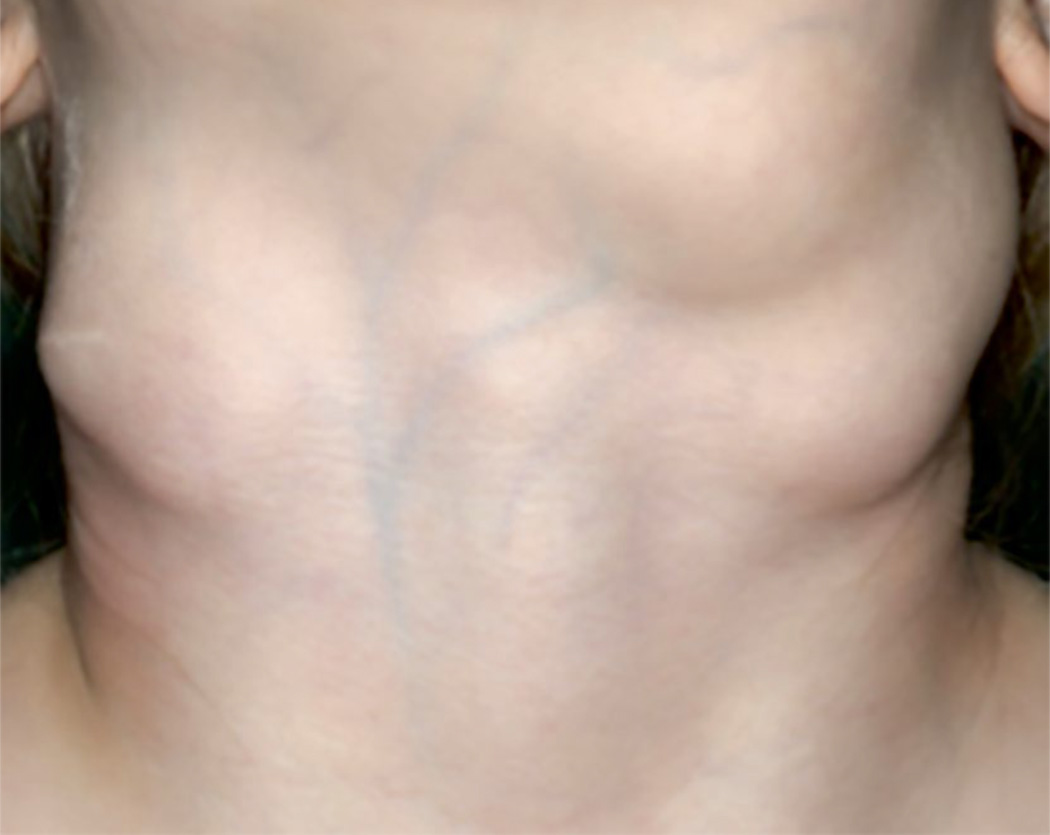
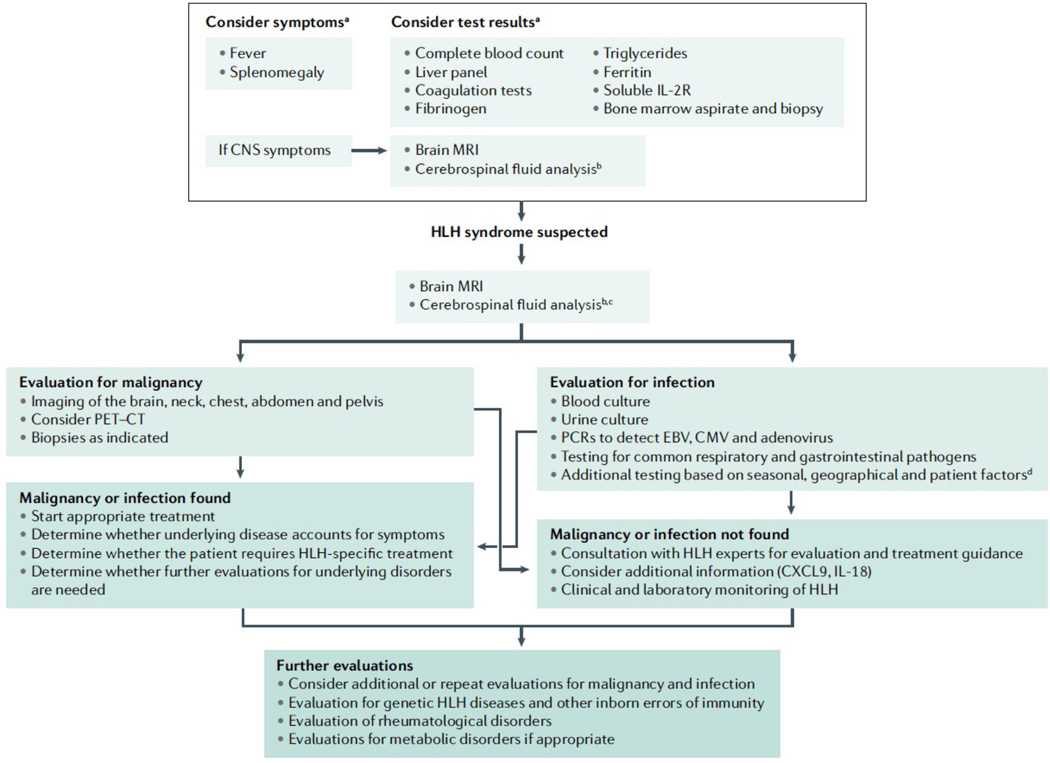
References
-
- Favara BE et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med. Pediatr. Oncol 29, 157–166 (1997). - PubMed
-
- Aschoff L. & Kiyono K. Frage der grossen Mononulearn. Folia Haematol 15, 383–390 (1913).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
