

CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models
- PMID: 34622231
- PMCID: PMC8484689
- DOI: 10.1016/j.xcrm.2021.100394
CCNE1 copy number is a biomarker for response to combination WEE1-ATR inhibition in ovarian and endometrial cancer models
Abstract
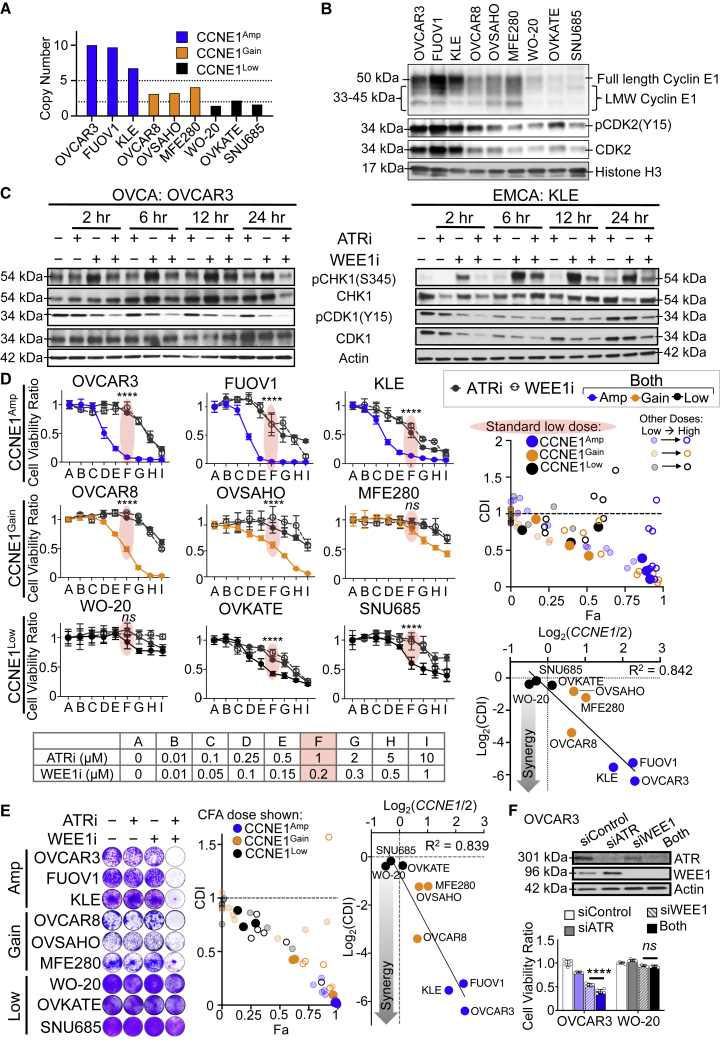
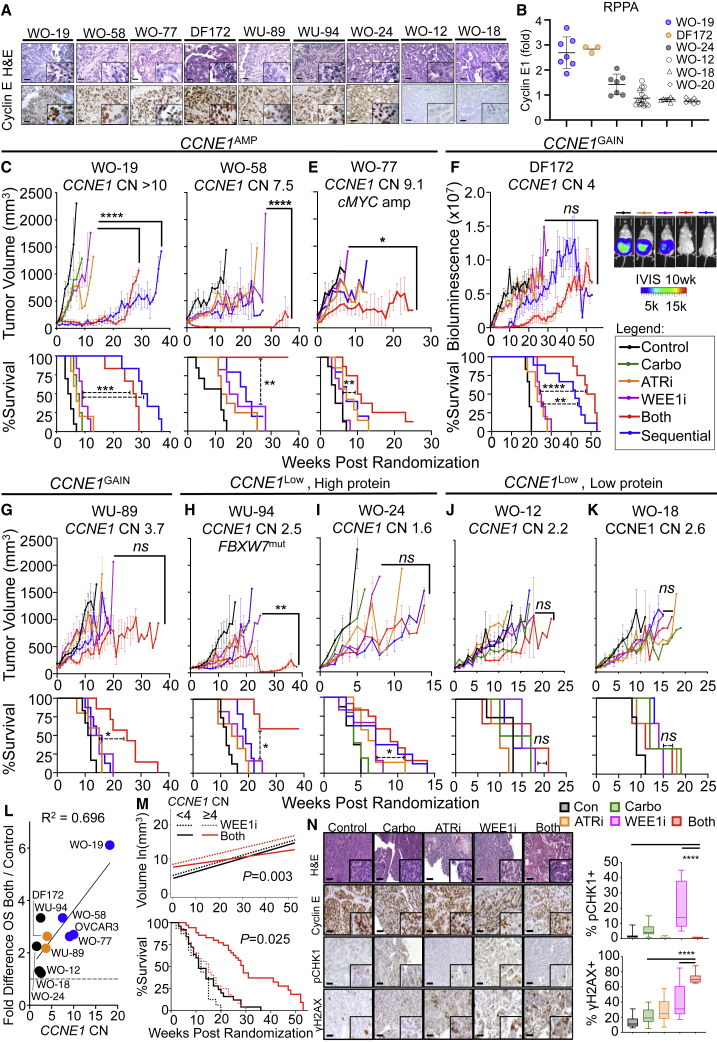
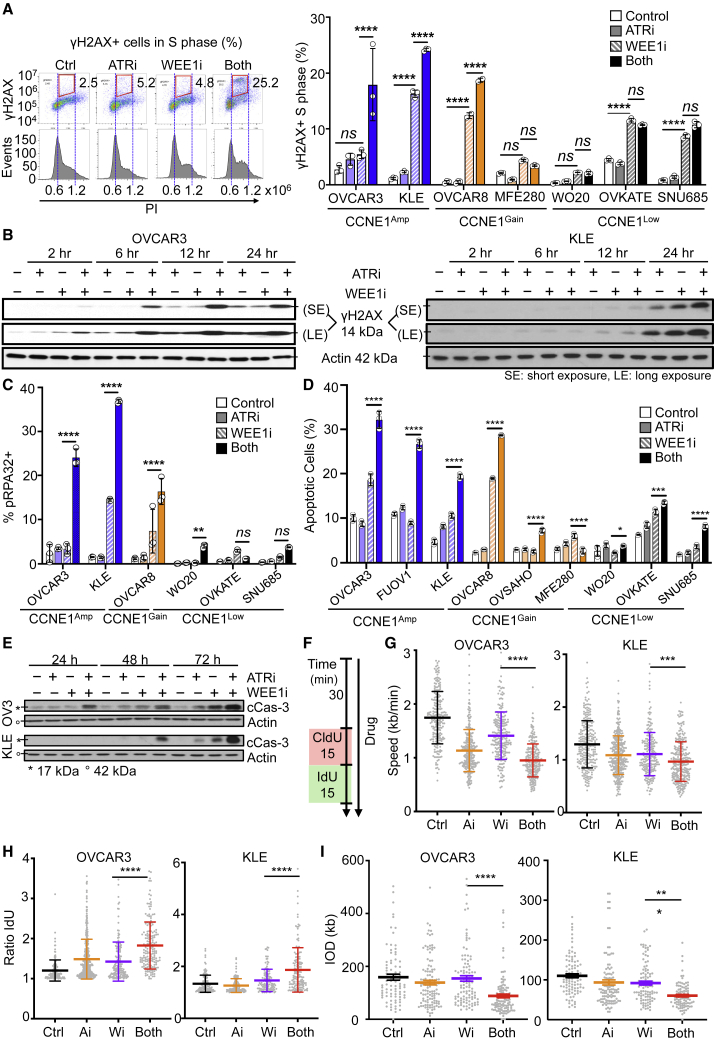
CCNE1-amplified ovarian cancers (OVCAs) and endometrial cancers (EMCAs) are associated with platinum resistance and poor survival, representing a clinically unmet need. We hypothesized that dysregulated cell-cycle progression promoted by CCNE1 overexpression would lead to increased sensitivity to low-dose WEE1 inhibition and ataxia telangiectasia and Rad3-related (ATR) inhibition (WEE1i-ATRi), thereby optimizing efficacy and tolerability. The addition of ATRi to WEE1i is required to block feedback activation of ATR signaling mediated by WEE1i. Low-dose WEE1i-ATRi synergistically decreases viability and colony formation and increases replication fork collapse and double-strand breaks (DSBs) in a CCNE1 copy number (CN)-dependent manner. Only upon CCNE1 induction does WEE1i perturb DNA synthesis at S-phase entry, and addition of ATRi increases DSBs during DNA synthesis. Inherent resistance to WEE1i is overcome with WEE1i-ATRi, with notable durable tumor regressions and improved survival in patient-derived xenograft (PDX) models in a CCNE1-level-dependent manner. These studies demonstrate that CCNE1 CN is a clinically tractable biomarker predicting responsiveness to low-dose WEE1i-ATRi for aggressive subsets of OVCAs/EMCAs.
Keywords: ATR; CCNE1 copy number; WEE1; biomarker; ovarian and endometrial cancer.
© 2021 The Author(s).
Conflict of interest statement
F.S. serves on a scientific advisory board and has received funding for clinical trials from AstraZeneca. G.B.M. receives support or acts as a consultant for AstraZeneca, ImmunoMET, Ionis, Nanostring, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, Abbvie, Amphista, Ellipses Pharma, Eli Lilly, Medacorp, Turbine, Zentalis Pharmaceuticals, and Tarveda and has transferred technology to Myriad and Nanostring. E.J.B. serves on the scientific advisory board of Atrin Pharmaceuticals and has been an advisor for Sierra Oncology. R.D. serves on the scientific advisory board of Repare Therapeutics and Siamab Therapeutics and advises Mersana Therapeutics and nVision Medical.
Figures





Comment in
-
WEE1i-ATRi combination therapy: a promising low-dose treatment for CCNE1-amplified gynecologic cancers.Cell Rep Med. 2021 Sep 23;2(9):100402. doi: 10.1016/j.xcrm.2021.100402. eCollection 2021 Sep 21. Cell Rep Med. 2021. PMID: 34622238 Free PMC article.
References
-
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. - PubMed
-
- Birrer, M.J., and Ceppi, L., eds. (2017). Translational Advances in Gynecologic Cancers (Academic Press).
-
- Armstrong D.K., Bundy B., Wenzel L., Huang H.Q., Baergen R., Lele S., Copeland L.J., Walker J.L., Burger R.A., Gynecologic Oncology Group Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006;354:34–43. - PubMed
-
- Winter W.E., 3rd, Maxwell G.L., Tian C., Carlson J.W., Ozols R.F., Rose P.G., Markman M., Armstrong D.K., Muggia F., McGuire W.P., Gynecologic Oncology Group Study Prognostic factors for stage III epithelial ovarian cancer: a Gynecologic Oncology Group Study. J. Clin. Oncol. 2007;25:3621–3627. - PubMed
-
- Patch A.M., Christie E.L., Etemadmoghadam D., Garsed D.W., George J., Fereday S., Nones K., Cowin P., Alsop K., Bailey P.J., Australian Ovarian Cancer Study Group Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
