

Clustering spatial transcriptomics data
- PMID: 34623423
- PMCID: PMC8796363
- DOI: 10.1093/bioinformatics/btab704
Clustering spatial transcriptomics data
Erratum in
-
Correction to: Clustering spatial transcriptomics data.Bioinformatics. 2023 Sep 2;39(9):btad574. doi: 10.1093/bioinformatics/btad574. Bioinformatics. 2023. PMID: 37738522 Free PMC article. No abstract available.
Abstract
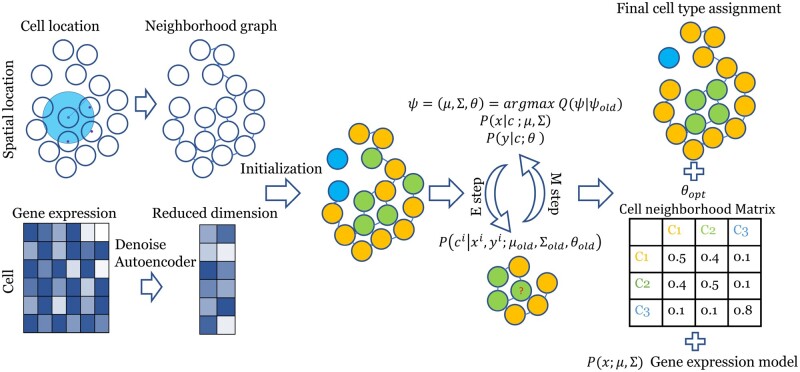
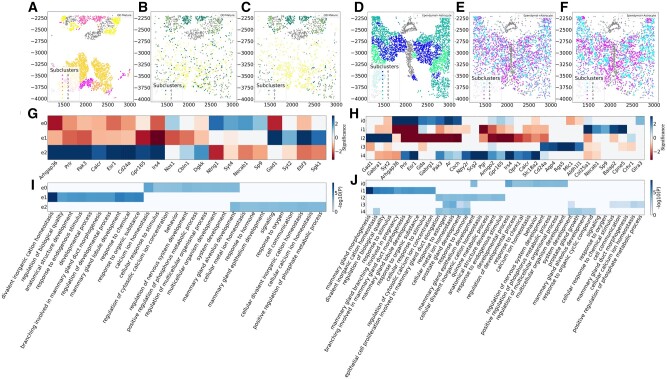
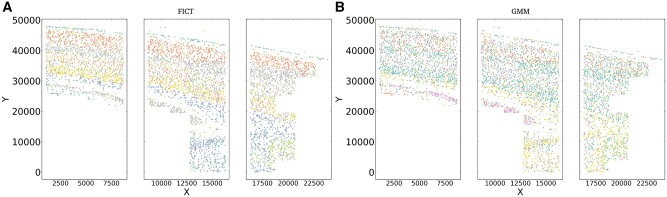
Motivation: Recent advancements in fluorescence in situ hybridization (FISH) techniques enable them to concurrently obtain information on the location and gene expression of single cells. A key question in the initial analysis of such spatial transcriptomics data is the assignment of cell types. To date, most studies used methods that only rely on the expression levels of the genes in each cell for such assignments. To fully utilize the data and to improve the ability to identify novel sub-types, we developed a new method, FICT, which combines both expression and neighborhood information when assigning cell types.
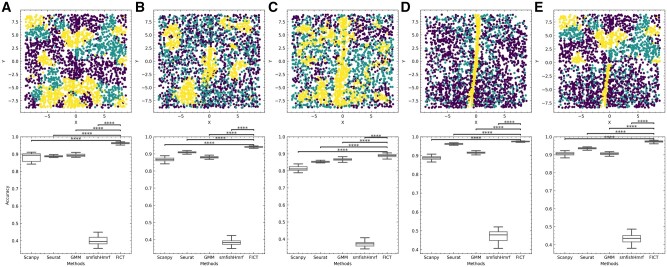
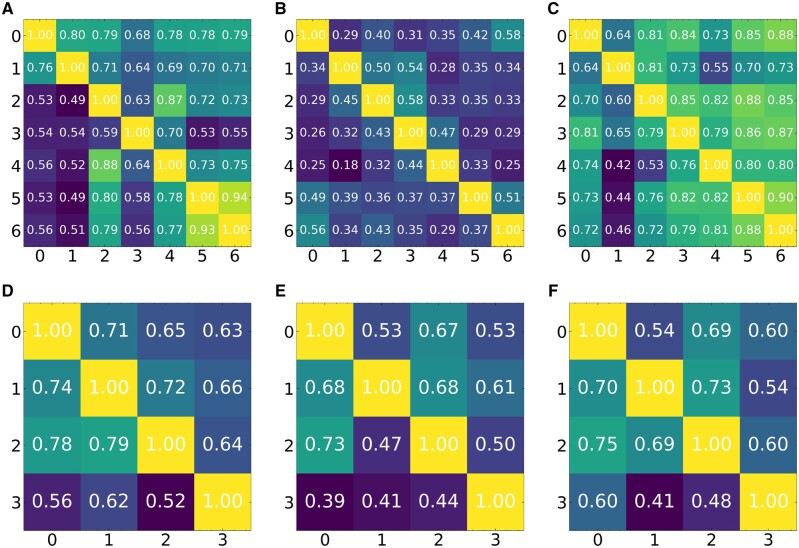
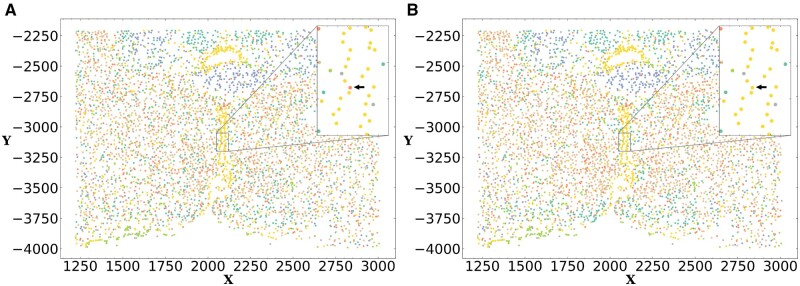
Results: FICT optimizes a probabilistic function that we formalize and for which we provide learning and inference algorithms. We used FICT to analyze both simulated and several real spatial transcriptomics data. As we show, FICT can accurately identify cell types and sub-types, improving on expression only methods and other methods proposed for clustering spatial transcriptomics data. Some of the spatial sub-types identified by FICT provide novel hypotheses about the new functions for excitatory and inhibitory neurons.
Availability and implementation: FICT is available at: https://github.com/haotianteng/FICT.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Besag J. (1986) On the statistical analysis of dirty pictures. J. R. Stat. Soc. Ser. B (Methodological), 48, 259–279.
-
- Blondel V.D. et al. (2008) Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp., 2008, P10008.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
