

Endocrine resistance in breast cancer: from molecular mechanisms to therapeutic strategies
- PMID: 34623477
- PMCID: PMC8611518
- DOI: 10.1007/s00109-021-02136-5
Endocrine resistance in breast cancer: from molecular mechanisms to therapeutic strategies
Abstract
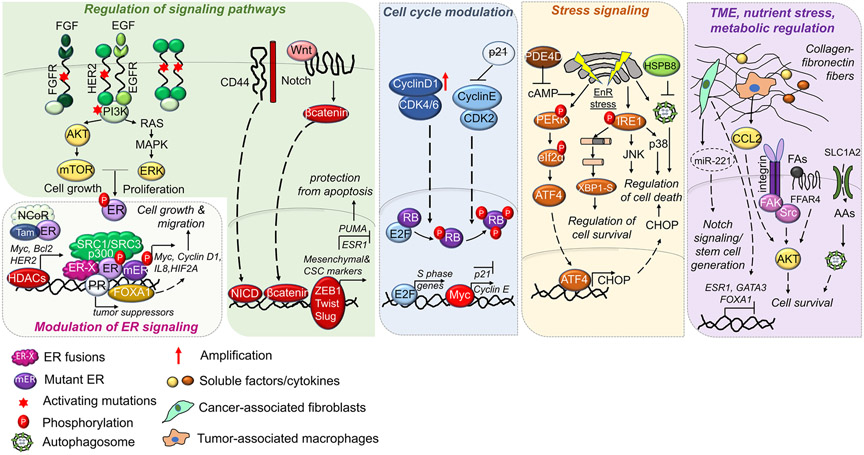
Estrogen receptor-positive (ER +) breast cancer accounts for approximately 75% of all breast cancers. Endocrine therapies, including selective ER modulators (SERMs), aromatase inhibitors (AIs), and selective ER down-regulators (SERDs) provide substantial clinical benefit by reducing the risk of disease recurrence and mortality. However, resistance to endocrine therapies represents a major challenge, limiting the success of ER + breast cancer treatment. Mechanisms of endocrine resistance involve alterations in ER signaling via modulation of ER (e.g., ER downregulation, ESR1 mutations or fusions); alterations in ER coactivators/corepressors, transcription factors (TFs), nuclear receptors and epigenetic modulators; regulation of signaling pathways; modulation of cell cycle regulators; stress signaling; and alterations in tumor microenvironment, nutrient stress, and metabolic regulation. Current therapeutic strategies to improve outcome of endocrine-resistant patients in clinics include inhibitors against mechanistic target of rapamycin (mTOR), cyclin-dependent kinase (CDK) 4/6, and the phosphoinositide 3-kinase (PI3K) subunit, p110α. Preclinical studies reveal novel therapeutic targets, some of which are currently tested in clinical trials as single agents or in combination with endocrine therapies, such as ER partial agonists, ER proteolysis targeting chimeras (PROTACs), next-generation SERDs, AKT inhibitors, epidermal growth factor receptor 1 and 2 (EGFR/HER2) dual inhibitors, HER2 targeting antibody-drug conjugates (ADCs) and histone deacetylase (HDAC) inhibitors. In this review, we summarize the established and emerging mechanisms of endocrine resistance, alterations during metastatic recurrence, and discuss the approved therapies and ongoing clinical trials testing the combination of novel targeted therapies with endocrine therapy in endocrine-resistant ER + breast cancer patients.
Keywords: Breast cancer; Endocrine resistance; Mechanisms of resistance; Therapy.
© 2021. The Author(s), under exclusive licence to Springer-Verlag GmbH Germany, part of Springer Nature.
Conflict of interest statement
Figures

References
-
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries (2018) CA: a cancer journal for clinicians 68: 394–424. - PubMed
-
- DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A et al. Breast cancer statistics, 2019 (2019) CA: a cancer journal for clinicians 69: 438–451. - PubMed
-
- Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer (2009) Nat Rev Cancer 9: 631–643. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
