

Haplotype heterogeneity and low linkage disequilibrium reduce reliable prediction of genotypes for the ‑α 3.7I form of α-thalassaemia using genome-wide microarray data
- PMID: 34632085
- PMCID: PMC8474104
- DOI: 10.12688/wellcomeopenres.16320.2
Haplotype heterogeneity and low linkage disequilibrium reduce reliable prediction of genotypes for the ‑α 3.7I form of α-thalassaemia using genome-wide microarray data
Abstract
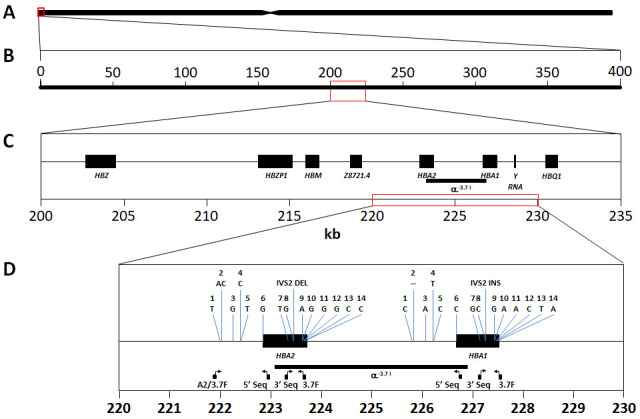
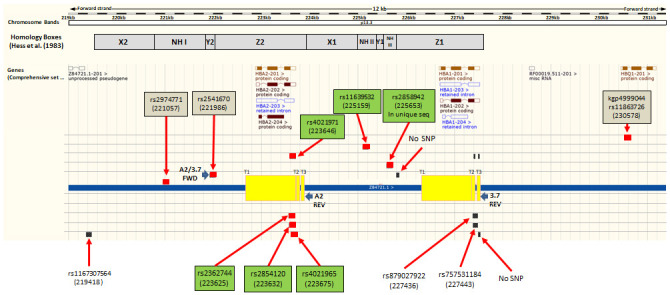
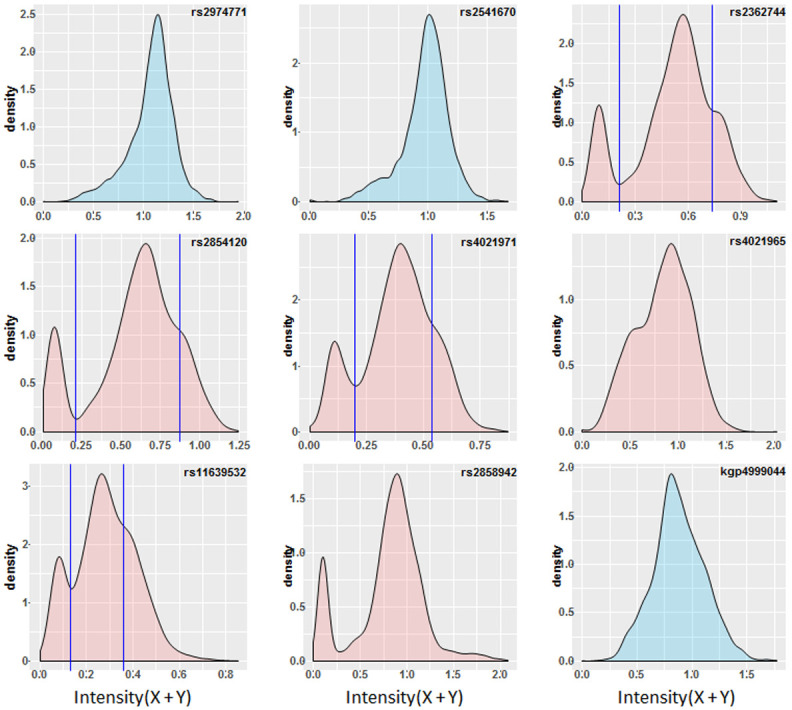
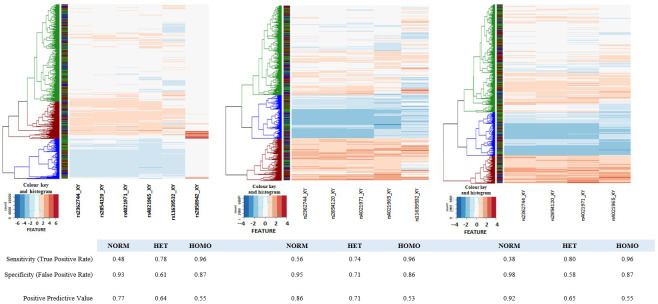
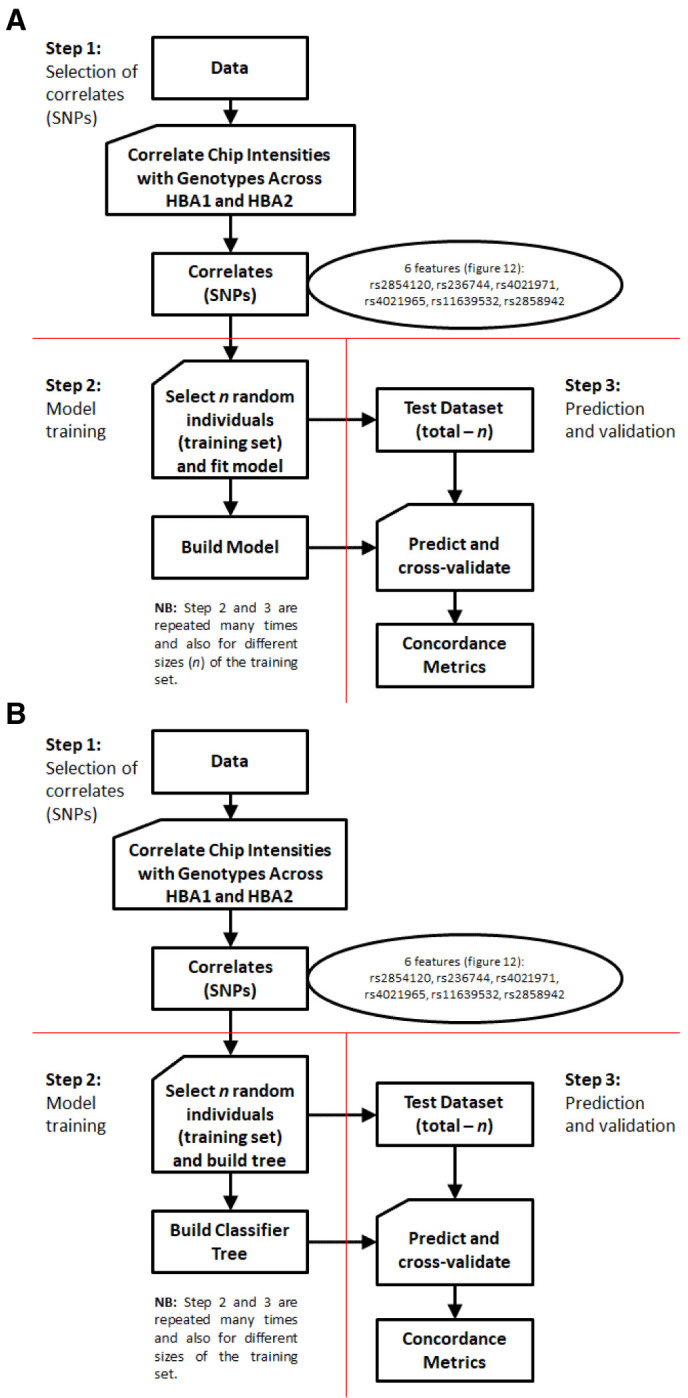
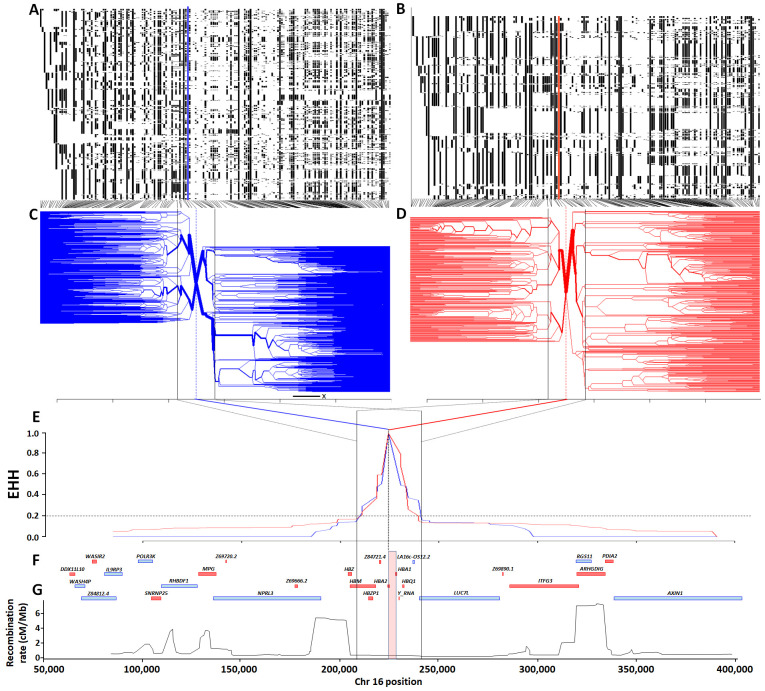
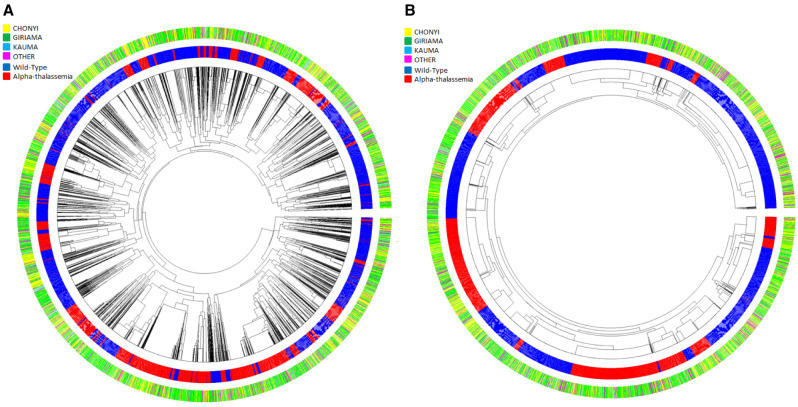
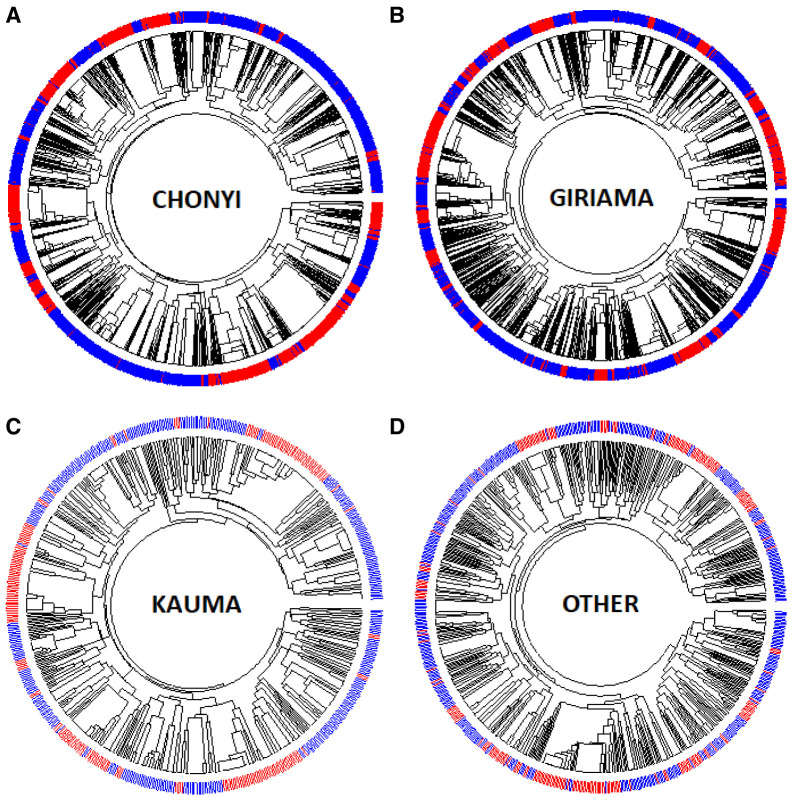
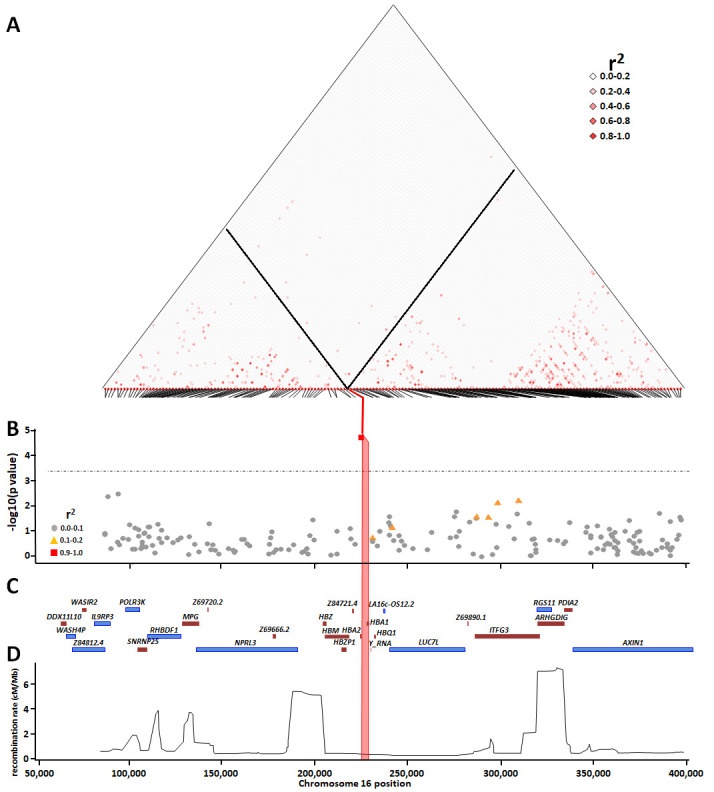
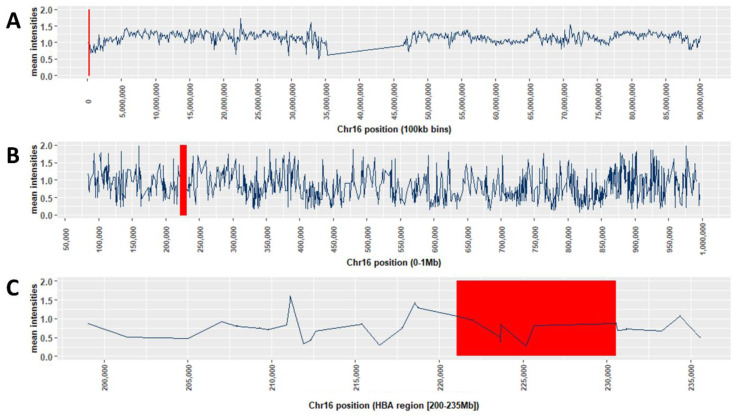
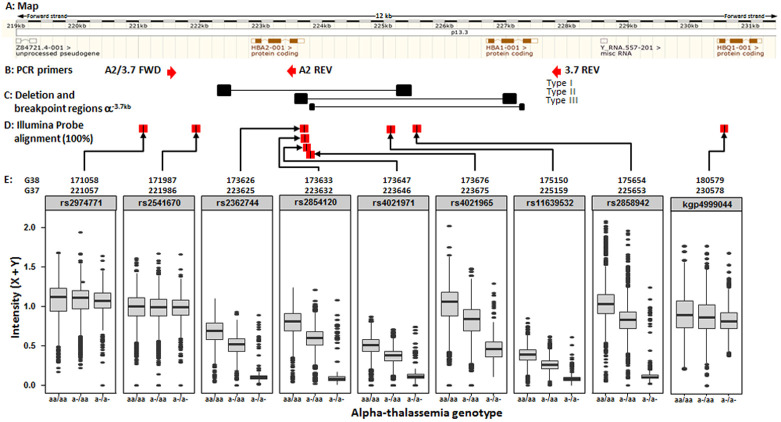
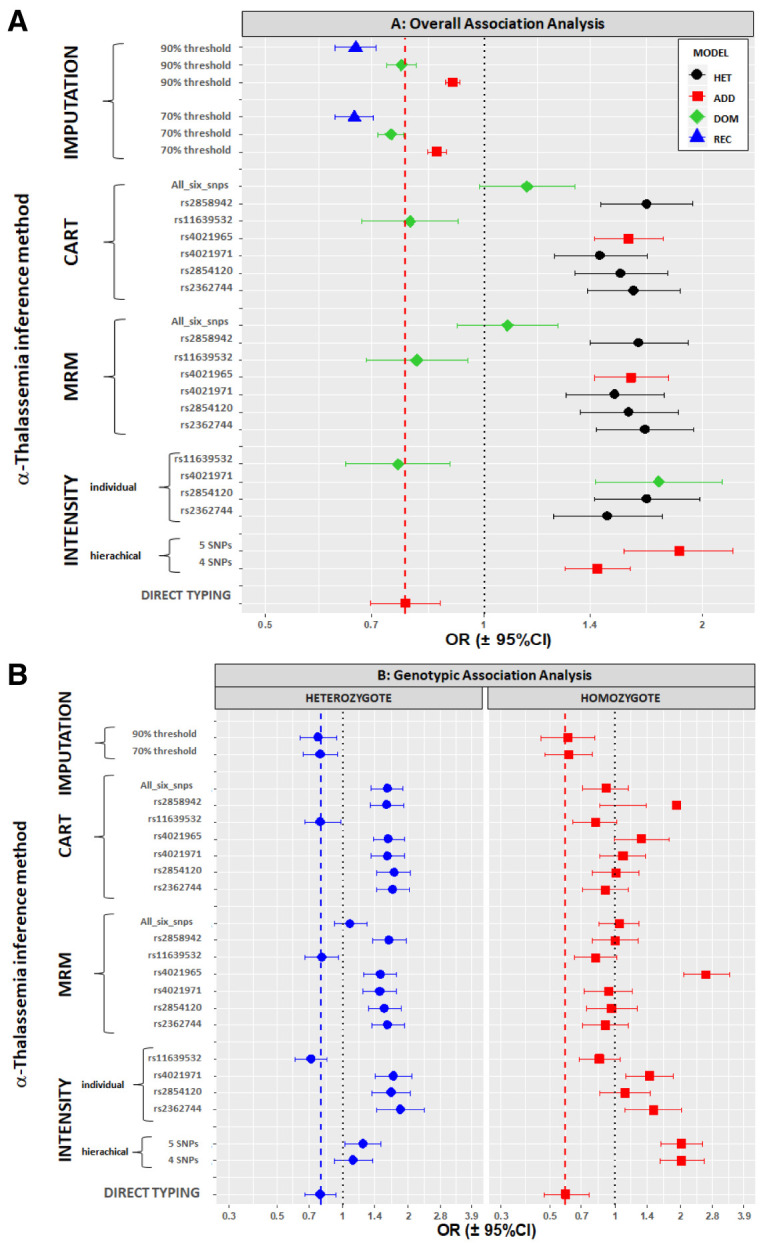
Background: The -α 3.7I-thalassaemia deletion is very common throughout Africa because it protects against malaria. When undertaking studies to investigate human genetic adaptations to malaria or other diseases, it is important to account for any confounding effects of α-thalassaemia to rule out spurious associations. Methods: In this study, we have used direct α-thalassaemia genotyping to understand why GWAS data from a large malaria association study in Kilifi Kenya did not identify the α-thalassaemia signal. We then explored the potential use of a number of new approaches to using GWAS data for imputing α-thalassaemia as an alternative to direct genotyping by PCR. Results: We found very low linkage-disequilibrium of the directly typed data with the GWAS SNP markers around α-thalassaemia and across the haemoglobin-alpha ( HBA) gene region, which along with a complex haplotype structure, could explain the lack of an association signal from the GWAS SNP data. Some indirect typing methods gave results that were in broad agreement with those derived from direct genotyping and could identify an association signal, but none were sufficiently accurate to allow correct interpretation compared with direct typing, leading to confusing or erroneous results. Conclusions: We conclude that going forwards, direct typing methods such as PCR will still be required to account for α-thalassaemia in GWAS studies.
Keywords: Classification and Regression Tree; GWAS; Malaria; Predictive Models; haplotypes; multinomial regression-model; α-thalassaemia.
Copyright: © 2021 Ndila CM et al.
Conflict of interest statement
No competing interests were disclosed.
Figures

References
-
- Weatherall DJ, Clegg JB: The thalassaemia syndromes. Blackwell Scientific Publications, Oxford,2002. 10.1002/9780470696705 - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
