

Modular self-assembly system for development of oligomeric, highly internalizing and potent cytotoxic conjugates targeting fibroblast growth factor receptors
- PMID: 34635096
- PMCID: PMC8504119
- DOI: 10.1186/s12929-021-00767-x
Modular self-assembly system for development of oligomeric, highly internalizing and potent cytotoxic conjugates targeting fibroblast growth factor receptors
Abstract
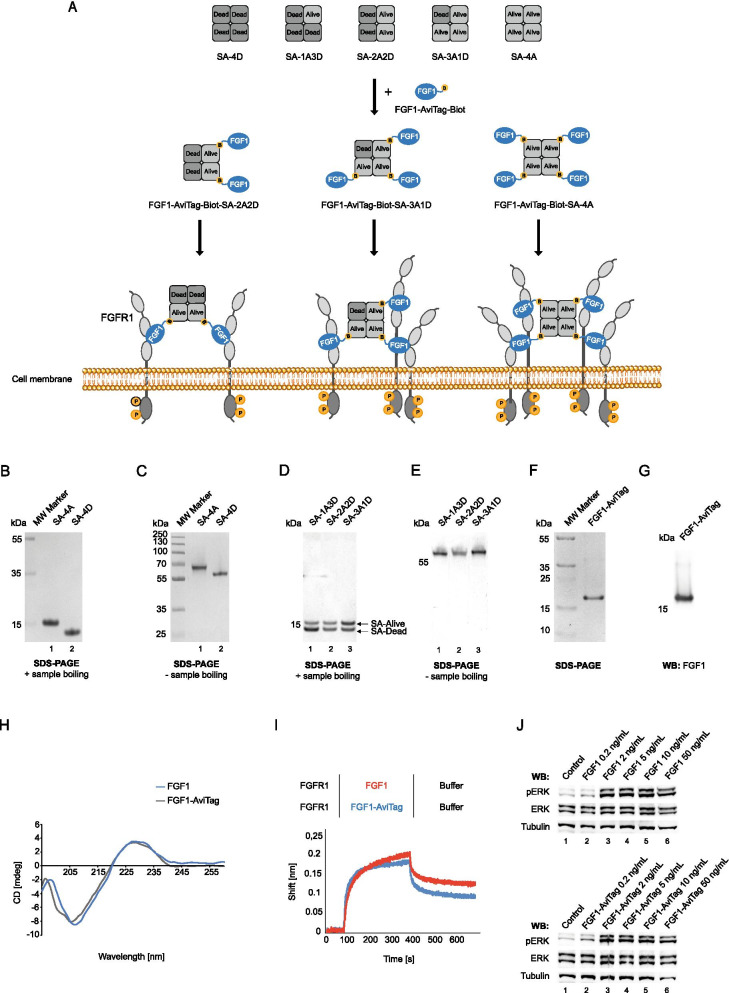
Background: Overexpression of FGFR1 is observed in numerous tumors and therefore this receptor constitutes an attractive molecular target for selective cancer treatment with cytotoxic conjugates. The success of cancer therapy with cytotoxic conjugates largely relies on the precise recognition of a cancer-specific marker by a targeting molecule within the conjugate and its subsequent cellular internalization by receptor mediated endocytosis. We have recently demonstrated that efficiency and mechanism of FGFR1 internalization are governed by spatial distribution of the receptor in the plasma membrane, where clustering of FGFR1 into larger oligomers stimulated fast and highly efficient uptake of the receptor by simultaneous engagement of multiple endocytic routes. Based on these findings we aimed to develop a modular, self-assembly system for generation of oligomeric cytotoxic conjugates, capable of FGFR1 clustering, for targeting FGFR1-overproducing cancer cells.
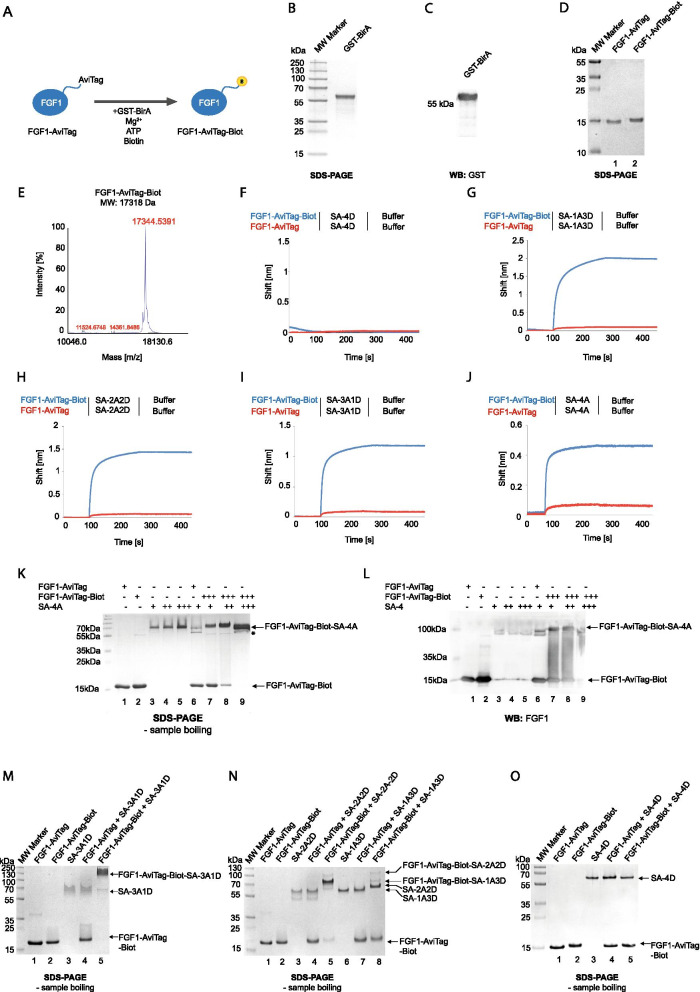
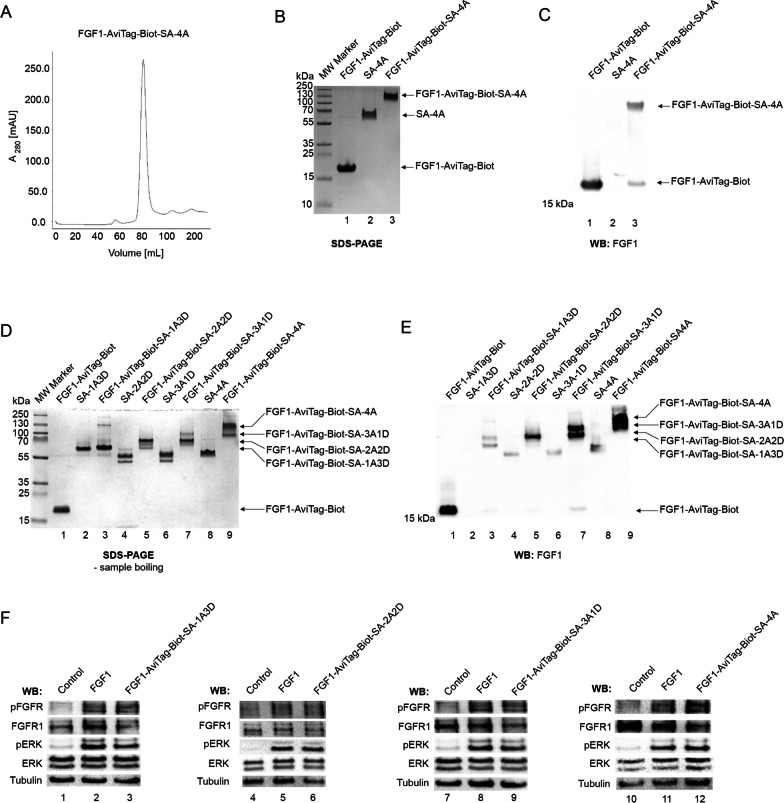
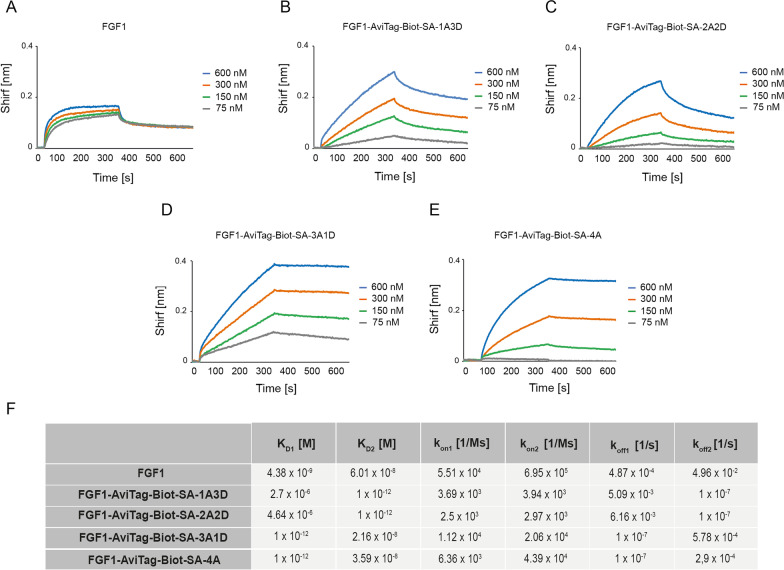
Methods: Engineered FGF1 was used as FGFR1-recognition molecule and tailored for enhanced stability and site-specific attachment of the cytotoxic drug. Modified streptavidin, allowing for controlled oligomerization of FGF1 variant was used for self-assembly of well-defined FGF1 oligomers of different valency and oligomeric cytotoxic conjugate. Protein biochemistry methods were applied to obtain highly pure FGF1 oligomers and the oligomeric cytotoxic conjugate. Diverse biophysical, biochemical and cell biology tests were used to evaluate FGFR1 binding, internalization and the cytotoxicity of obtained oligomers.
Results: Developed multivalent FGF1 complexes are characterized by well-defined architecture, enhanced FGFR1 binding and improved cellular uptake. This successful strategy was applied to construct tetrameric cytotoxic conjugate targeting FGFR1-producing cancer cells. We have shown that enhanced affinity for the receptor and improved internalization result in a superior cytotoxicity of the tetrameric conjugate compared to the monomeric one.
Conclusions: Our data implicate that oligomerization of the targeting molecules constitutes an attractive strategy for improvement of the cytotoxicity of conjugates recognizing cancer-specific biomarkers. Importantly, the presented approach can be easily adapted for other tumor markers.
Keywords: Cancer; Endocytosis; FGF/FGFR; Targeted therapy.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Intrinsically Fluorescent Oligomeric Cytotoxic Conjugates Toxic for FGFR1-Overproducing Cancers.Biomacromolecules. 2021 Dec 13;22(12):5349-5362. doi: 10.1021/acs.biomac.1c01280. Epub 2021 Dec 2. Biomacromolecules. 2021. PMID: 34855396 Free PMC article.
-
The cytotoxic conjugate of highly internalizing tetravalent antibody for targeting FGFR1-overproducing cancer cells.Mol Med. 2021 May 7;27(1):46. doi: 10.1186/s10020-021-00306-2. Mol Med. 2021. PMID: 33962559 Free PMC article.
-
FGF1 Fusions with the Fc Fragment of IgG1 for the Assembly of GFPpolygons-Mediated Multivalent Complexes Recognizing FGFRs.Biomolecules. 2021 Jul 23;11(8):1088. doi: 10.3390/biom11081088. Biomolecules. 2021. PMID: 34439755 Free PMC article.
-
FGFR1 clustering with engineered tetravalent antibody improves the efficiency and modifies the mechanism of receptor internalization.Mol Oncol. 2020 Sep;14(9):1998-2021. doi: 10.1002/1878-0261.12740. Epub 2020 Jul 3. Mol Oncol. 2020. PMID: 32511887 Free PMC article.
-
Guided molecular missiles for tumor-targeting chemotherapy--case studies using the second-generation taxoids as warheads.Acc Chem Res. 2008 Jan;41(1):108-19. doi: 10.1021/ar700093f. Epub 2007 Jul 31. Acc Chem Res. 2008. PMID: 17663526 Review.
Cited by
-
Intrinsically Fluorescent Oligomeric Cytotoxic Conjugates Toxic for FGFR1-Overproducing Cancers.Biomacromolecules. 2021 Dec 13;22(12):5349-5362. doi: 10.1021/acs.biomac.1c01280. Epub 2021 Dec 2. Biomacromolecules. 2021. PMID: 34855396 Free PMC article.
-
The diverse dependence of galectin-1 and -8 on multivalency for the modulation of FGFR1 endocytosis.Cell Commun Signal. 2024 May 15;22(1):270. doi: 10.1186/s12964-024-01661-3. Cell Commun Signal. 2024. PMID: 38750548 Free PMC article.
-
The Significance of Cell Surface N-Glycosylation for Internalization and Potency of Cytotoxic Conjugates Targeting Receptor Tyrosine Kinases.Int J Mol Sci. 2022 Jul 31;23(15):8514. doi: 10.3390/ijms23158514. Int J Mol Sci. 2022. PMID: 35955648 Free PMC article.
-
Reprogramming FGF1 from the natural growth factor to the engineered heparan sulphate biosensor.Cell Commun Signal. 2025 May 28;23(1):248. doi: 10.1186/s12964-025-02269-x. Cell Commun Signal. 2025. PMID: 40437501 Free PMC article.
-
Di-caffeoylquinic acid: a potential inhibitor for amyloid-beta aggregation.J Nat Med. 2024 Sep;78(4):1029-1043. doi: 10.1007/s11418-024-01825-y. Epub 2024 Jun 27. J Nat Med. 2024. PMID: 38926328
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
