

Juvenile-Onset Huntington Disease Pathophysiology and Neurodevelopment: A Review
- PMID: 34636452
- PMCID: PMC9291924
- DOI: 10.1002/mds.28823
Juvenile-Onset Huntington Disease Pathophysiology and Neurodevelopment: A Review
Abstract
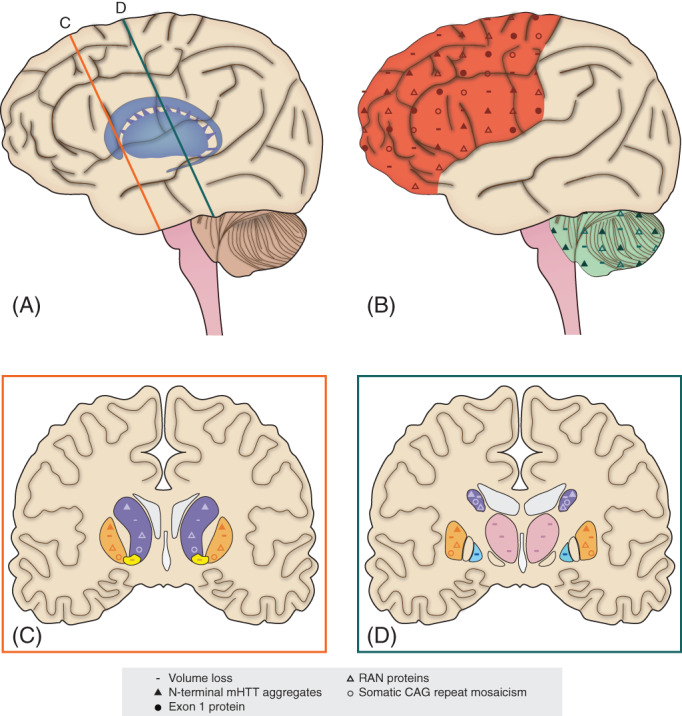
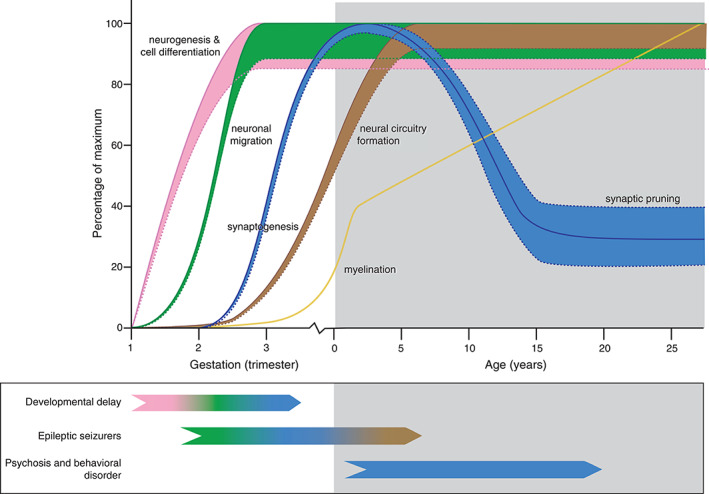
Huntington disease is an autosomal dominant inherited brain disorder that typically becomes manifest in adulthood. Juvenile-onset Huntington disease refers to approximately 5% of patients with symptom onset before the age of 21 years. The causal factor is a pathologically expanded CAG repeat in the Huntingtin gene. Age at onset is inversely correlated with CAG repeat length. Juvenile-onset patients have distinct symptoms and signs with more severe pathology of involved brain structures in comparison with disease onset in adulthood. The aim of this review is to compare clinical and pathological features in juvenile- and adult-onset Huntington disease and to explore which processes potentially contribute to the observed differences. A specific focus is placed on molecular mechanisms of mutant huntingtin in early neurodevelopment and the interaction of a neurodegenerative disease and postnatal brain maturation. The importance of a better understanding of pathophysiological differences between juvenile- and adult-onset Huntington disease lies in development and implementation of new therapeutic strategies. © 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Keywords: disease mechanisms; juvenile Huntington disease; neurodevelopment; pediatric Huntington disease; pediatric neurodegenerative disease.
© 2021 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Figures
References
-
- The Huntington Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington disease chromosomes. Cell 1993;72(6):971–983. - PubMed
-
- Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarrell O. Juvenile Huntington disease: does a dosage‐effect pathogenic mechanism differ from the classical adult disease? Mech Ageing Dev 2006;127(2):208–212. - PubMed
-
- Milunsky JM, Maher TA, Loose BA, Darras BT, Ito M. XL PCR for the detection of large trinucleotide expansions in juvenile Huntington disease. Clin Genet 2003;64(1):70–73. - PubMed
-
- Warren JD, Firgaira F, Thompson EM, Kneebone CS, Blumbergs PC, Thompson PD. The causes of sporadic and ‘senile’ chorea. Aust N Z J Med 1998;28(4):429–431. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
