

Interferon-γ Impairs Human Coronary Artery Endothelial Glucose Metabolism by Tryptophan Catabolism and Activates Fatty Acid Oxidation
- PMID: 34636650
- PMCID: PMC8595869
- DOI: 10.1161/CIRCULATIONAHA.121.053960
Interferon-γ Impairs Human Coronary Artery Endothelial Glucose Metabolism by Tryptophan Catabolism and Activates Fatty Acid Oxidation
Abstract
Background: Endothelial cells depend on glycolysis for much of their energy production. Impaired endothelial glycolysis has been associated with various vascular pathobiologies, including impaired angiogenesis and atherogenesis. IFN-γ (interferon-γ)-producing CD4+ and CD8+ T lymphocytes have been identified as the predominant pathological cell subsets in human atherosclerotic plaques. Although the immunologic consequences of these cells have been extensively evaluated, their IFN-γ-mediated metabolic effects on endothelial cells remain unknown. The purpose of this study was to determine the metabolic consequences of the T-lymphocyte cytokine, IFN-γ, on human coronary artery endothelial cells.
Methods: The metabolic effects of IFN-γ on primary human coronary artery endothelial cells were assessed by unbiased transcriptomic and metabolomic analyses combined with real-time extracellular flux analyses and molecular mechanistic studies. Cellular phenotypic correlations were made by measuring altered endothelial intracellular cGMP content, wound-healing capacity, and adhesion molecule expression.
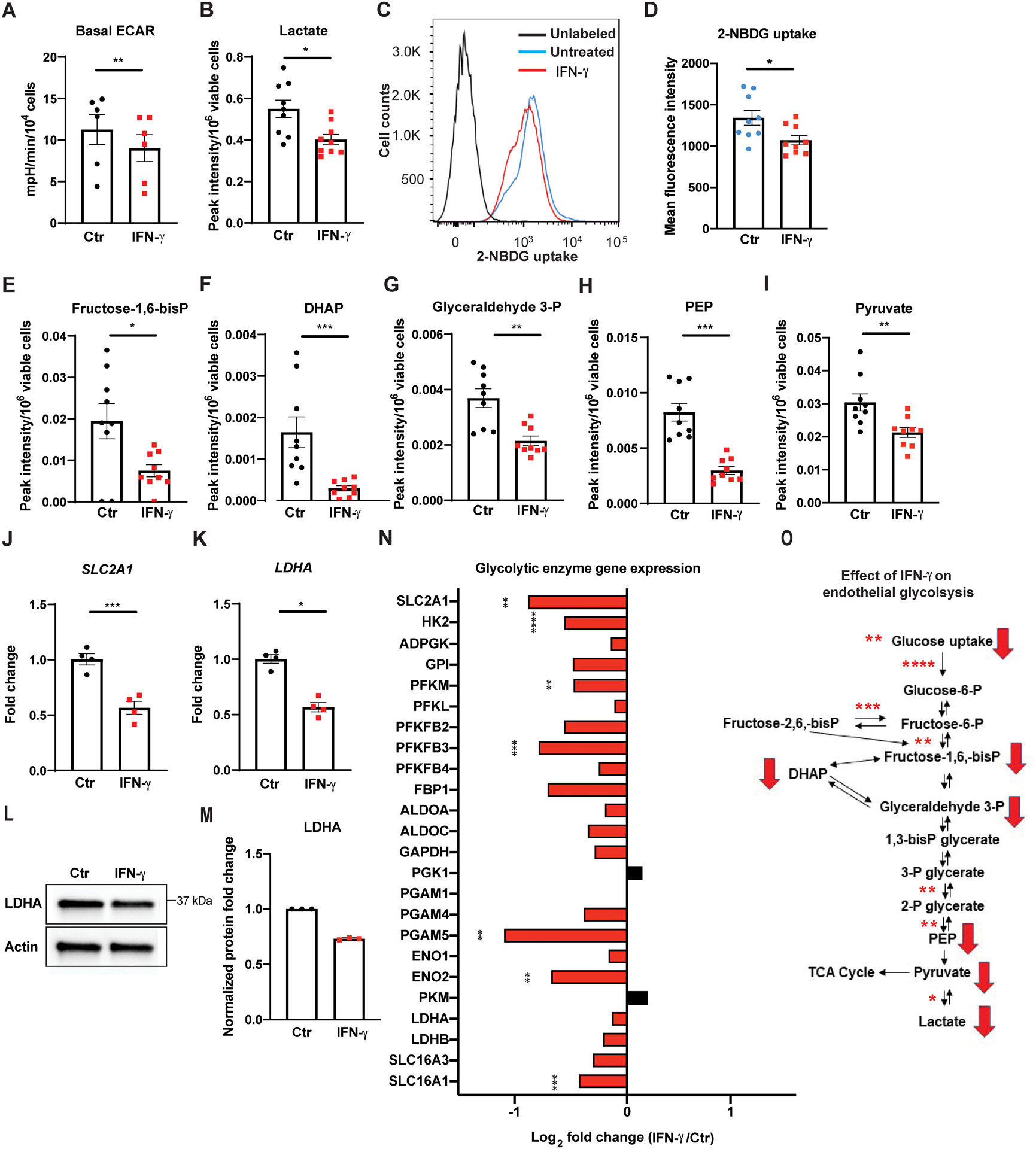
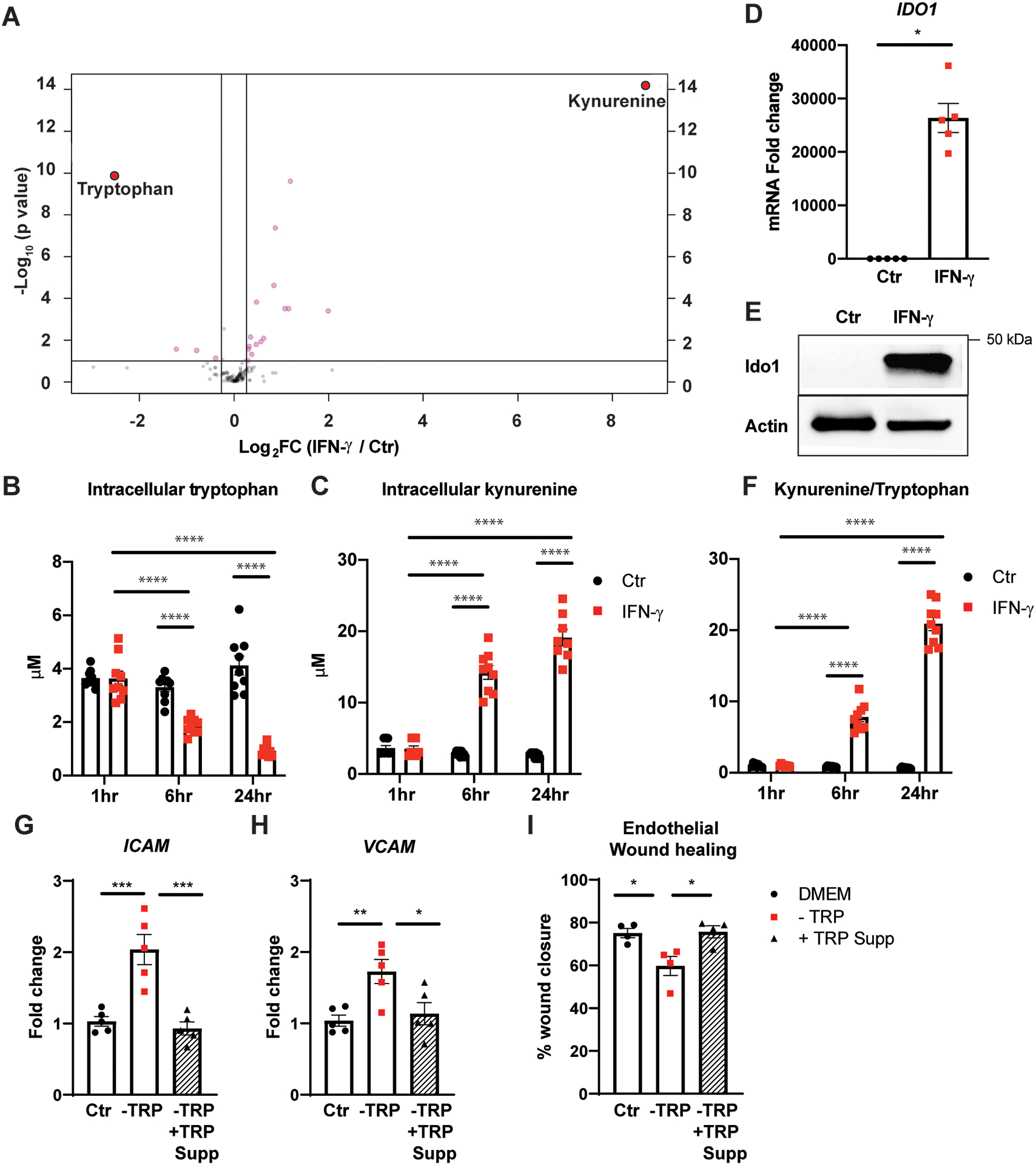
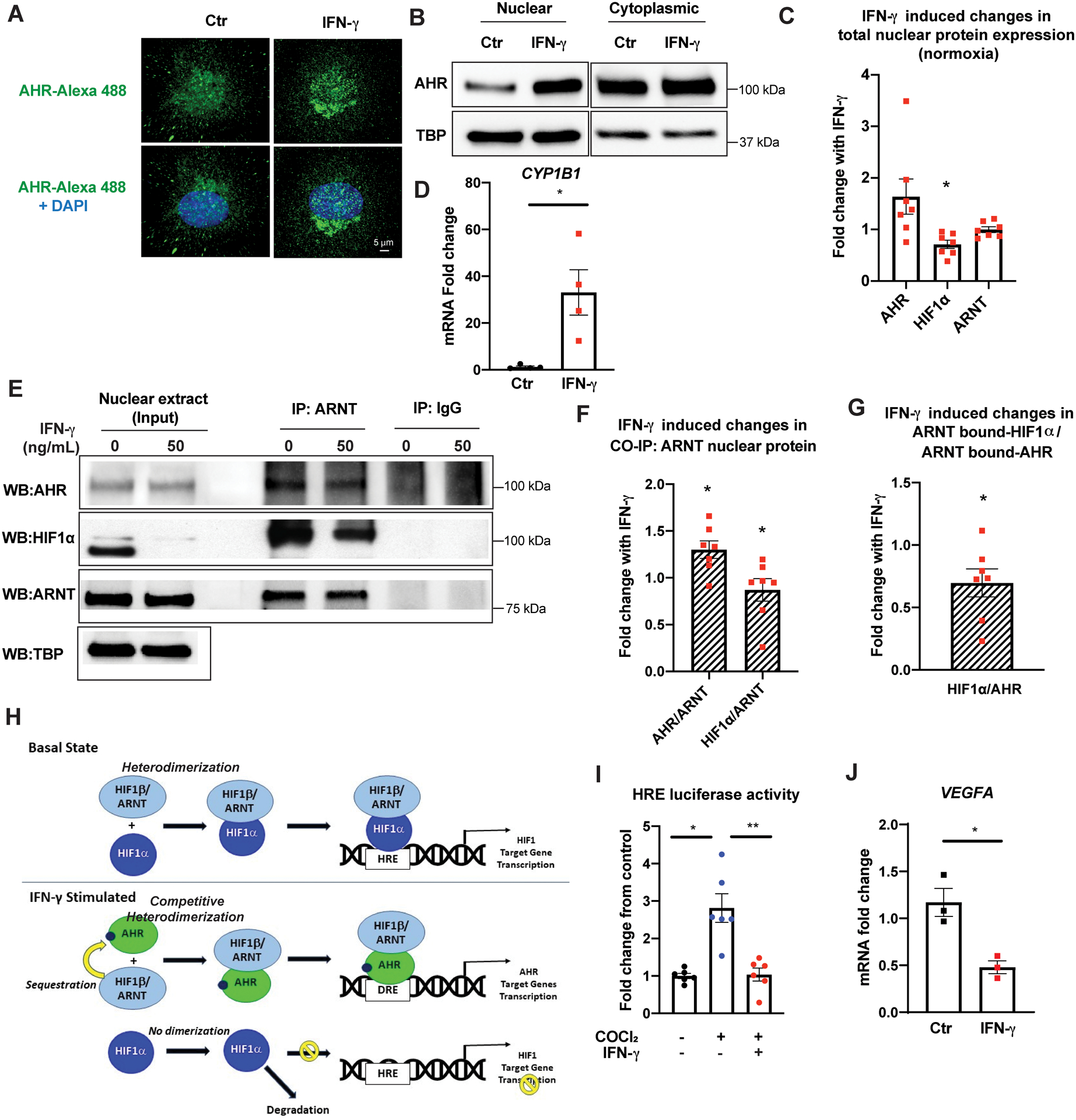
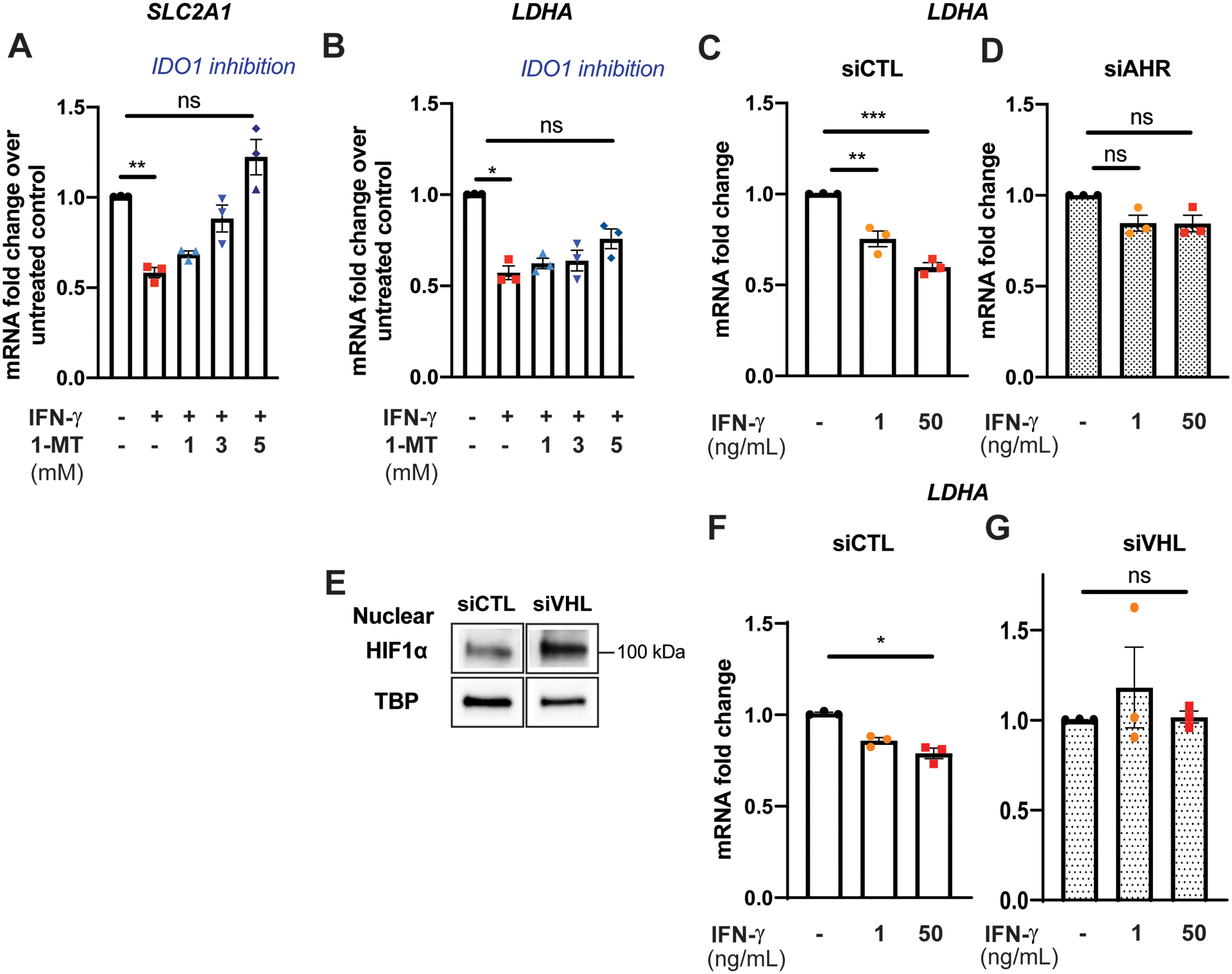
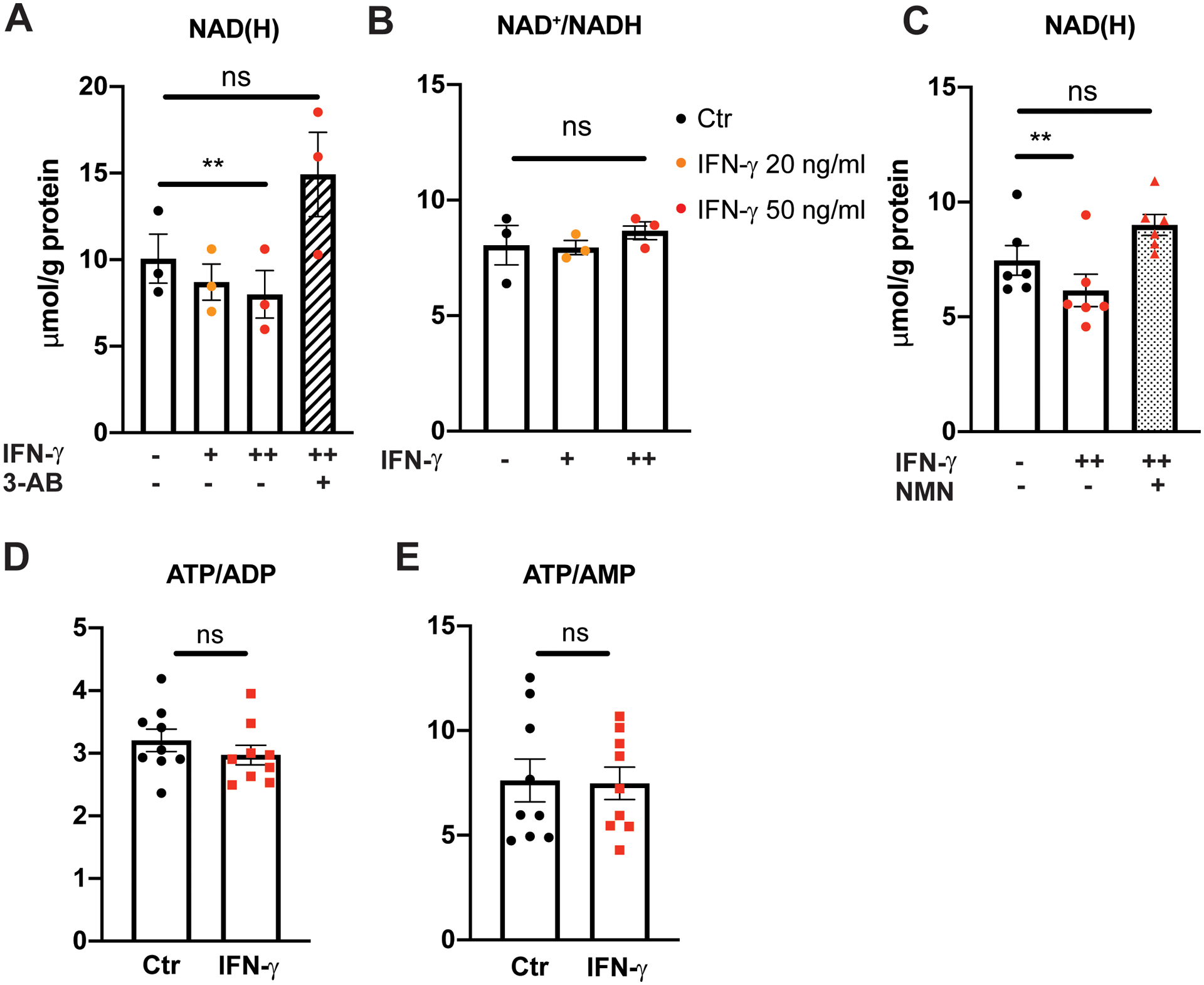
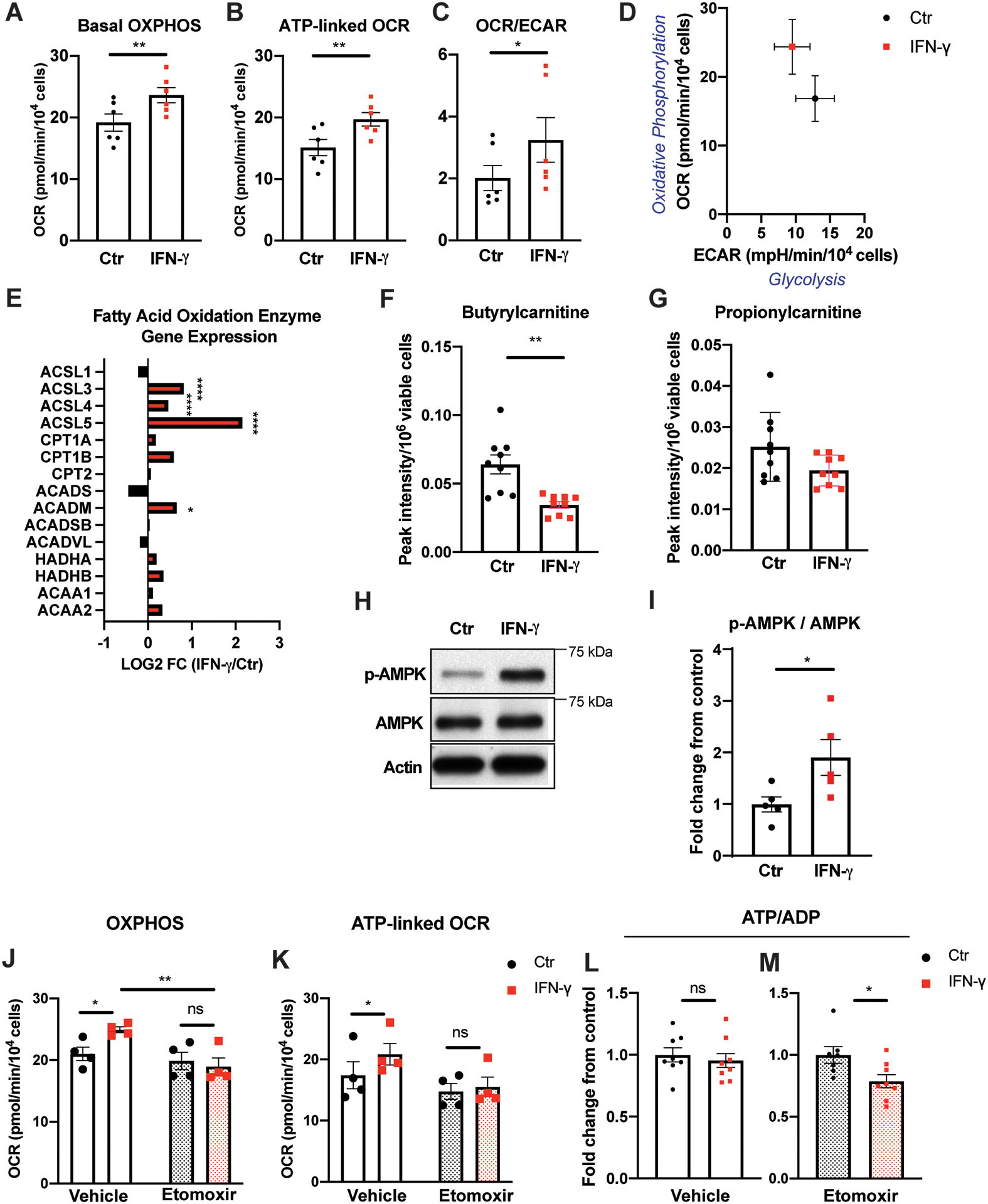
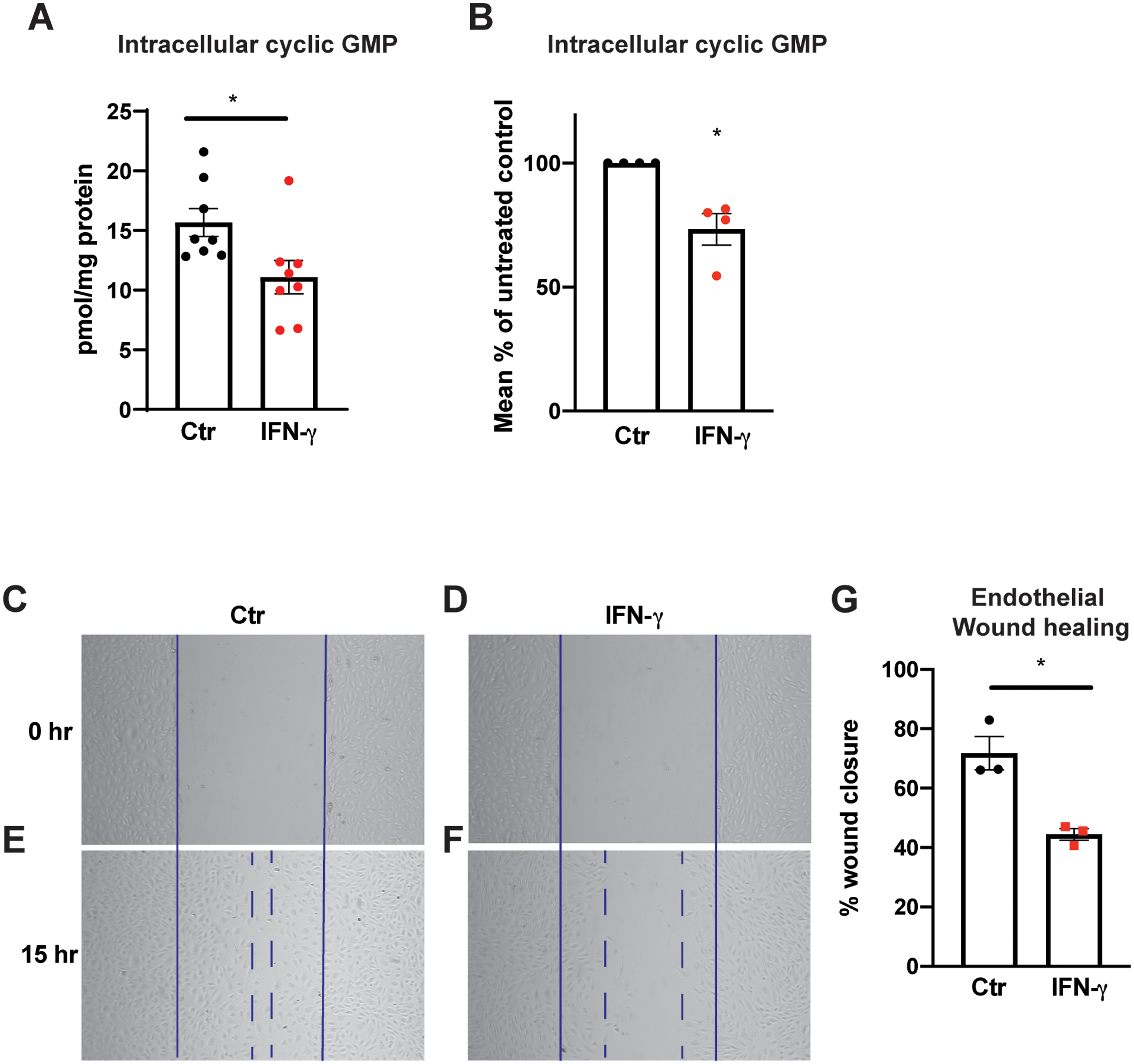
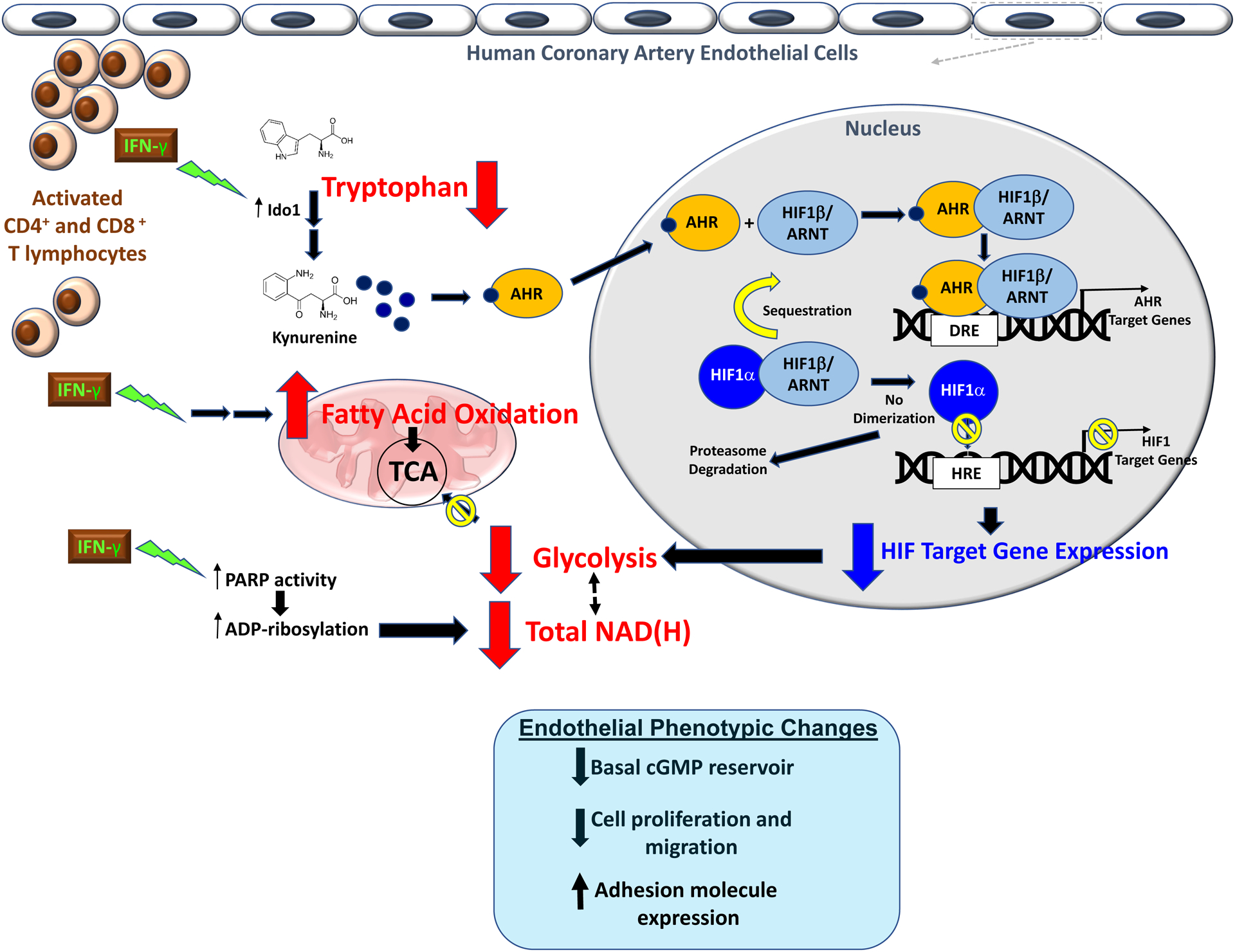
Results: IFN-γ exposure inhibited basal glycolysis of quiescent primary human coronary artery endothelial cells by 20% through the global transcriptional suppression of glycolytic enzymes resulting from decreased basal HIF1α (hypoxia-inducible factor 1α) nuclear availability in normoxia. The decrease in HIF1α activity was a consequence of IFN-γ-induced tryptophan catabolism resulting in ARNT (aryl hydrocarbon receptor nuclear translocator)/HIF1β sequestration by the kynurenine-activated AHR (aryl hydrocarbon receptor). In addition, IFN-γ resulted in a 23% depletion of intracellular nicotinamide adenine dinucleotide in human coronary artery endothelial cells. This altered glucose metabolism was met with concomitant activation of fatty acid oxidation, which augmented its contribution to intracellular ATP balance by >20%. These metabolic derangements were associated with adverse endothelial phenotypic changes, including decreased basal intracellular cGMP, impaired endothelial migration, and a switch to a proinflammatory state.
Conclusions: IFN-γ impairs endothelial glucose metabolism by altered tryptophan catabolism destabilizing HIF1, depletes nicotinamide adenine dinucleotide, and results in a metabolic shift toward increased fatty acid oxidation. This work suggests a novel mechanistic basis for pathological T lymphocyte-endothelial interactions in atherosclerosis mediated by IFN-γ, linking endothelial glucose, tryptophan, and fatty acid metabolism with the nicotinamide adenine dinucleotide balance and ATP generation and their adverse endothelial functional consequences.
Keywords: coronary artery; endothelium; fatty acid oxidation; glycolysis; interferon-γ; metabolism; tryptophan.
Figures
References
-
- Li X, Sun X and Carmeliet P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metabolism. 2019;30:414–433. - PubMed
-
- Krützfeldt A, Spahr R, Mertens S, Siegmund B and Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol. 1990;22:1393–404. - PubMed
-
- Culic O, Gruwel ML and Schrader J. Energy turnover of vascular endothelial cells. Am J Physiol. 1997;273:C205–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
