

CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial
- PMID: 34642489
- PMCID: PMC8516648
- DOI: 10.1038/s41591-021-01497-1
CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial
Abstract
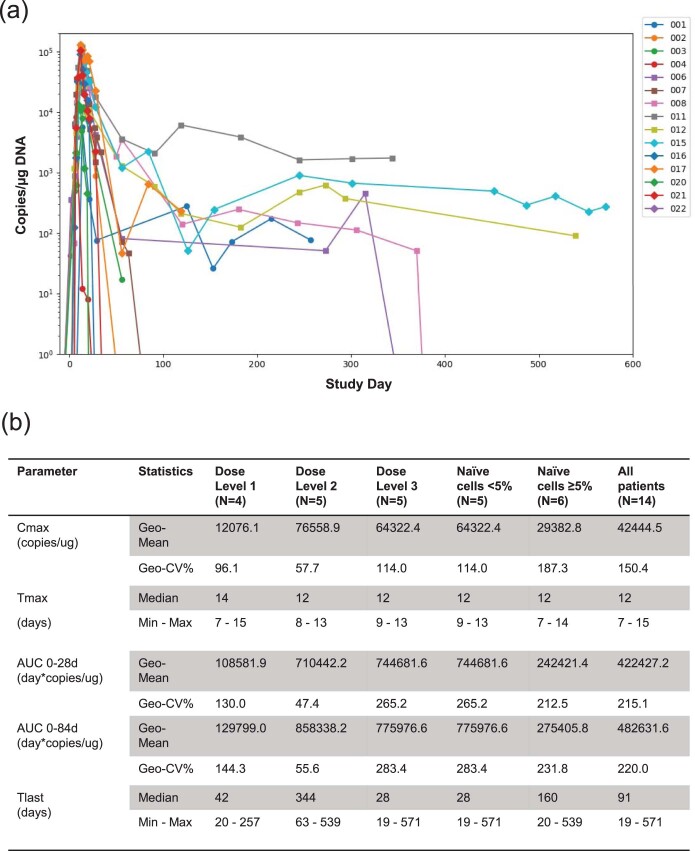
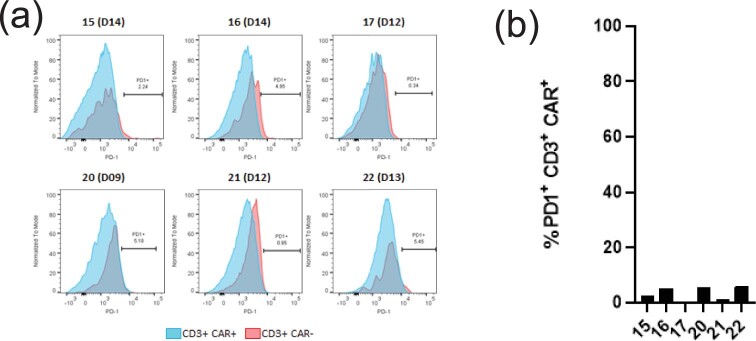
Chimeric antigen receptor (CAR) T cells targeting CD19 or CD22 have shown remarkable activity in B cell acute lymphoblastic leukemia (B-ALL). The major cause of treatment failure is antigen downregulation or loss. Dual antigen targeting could potentially prevent this, but the clinical safety and efficacy of CAR T cells targeting both CD19 and CD22 remain unclear. We conducted a phase 1 trial in pediatric and young adult patients with relapsed or refractory B-ALL (n = 15) to test AUTO3, autologous transduced T cells expressing both anti-CD19 and anti-CD22 CARs (AMELIA trial, EUDRA CT 2016-004680-39). The primary endpoints were the incidence of grade 3-5 toxicity in the dose-limiting toxicity period and the frequency of dose-limiting toxicities. Secondary endpoints included the rate of morphological remission (complete response or complete response with incomplete bone marrow recovery) with minimal residual disease-negative response, as well as the frequency and severity of adverse events, expansion and persistence of AUTO3, duration of B cell aplasia, and overall and event-free survival. The study endpoints were met. AUTO3 showed a favorable safety profile, with no dose-limiting toxicities or cases of AUTO3-related severe cytokine release syndrome or neurotoxicity reported. At 1 month after treatment the remission rate (that is, complete response or complete response with incomplete bone marrow recovery) was 86% (13 of 15 patients). The 1 year overall and event-free survival rates were 60% and 32%, respectively. Relapses were probably due to limited long-term AUTO3 persistence. Strategies to improve CAR T cell persistence are needed to fully realize the potential of dual targeting CAR T cell therapy in B-ALL.
© 2021. The Author(s).
Conflict of interest statement
F.F. has founder shares in Autolus. N.Z.K., V.G.R.P., K.S. and C.P. are shareholders in Autolus PLC. S.C., S.O., S.T., D.S.P., L.C., M.A.-H., M.M.F., V.B., M.F., S.S., R.J., F.A.V., K.D., W.D., Y.Z. and M.P. are shareholders in and employees of Autolus PLC. All other authors have no competing interests.
Figures
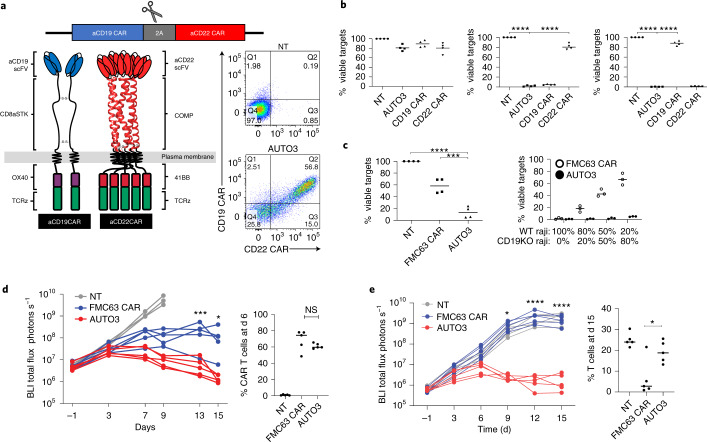
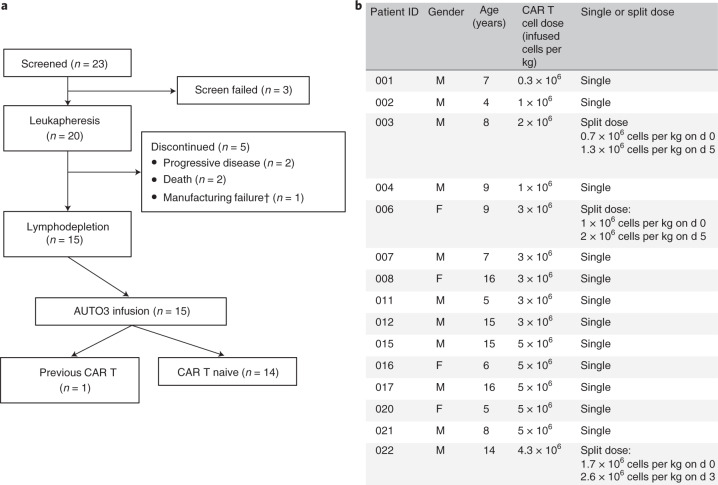

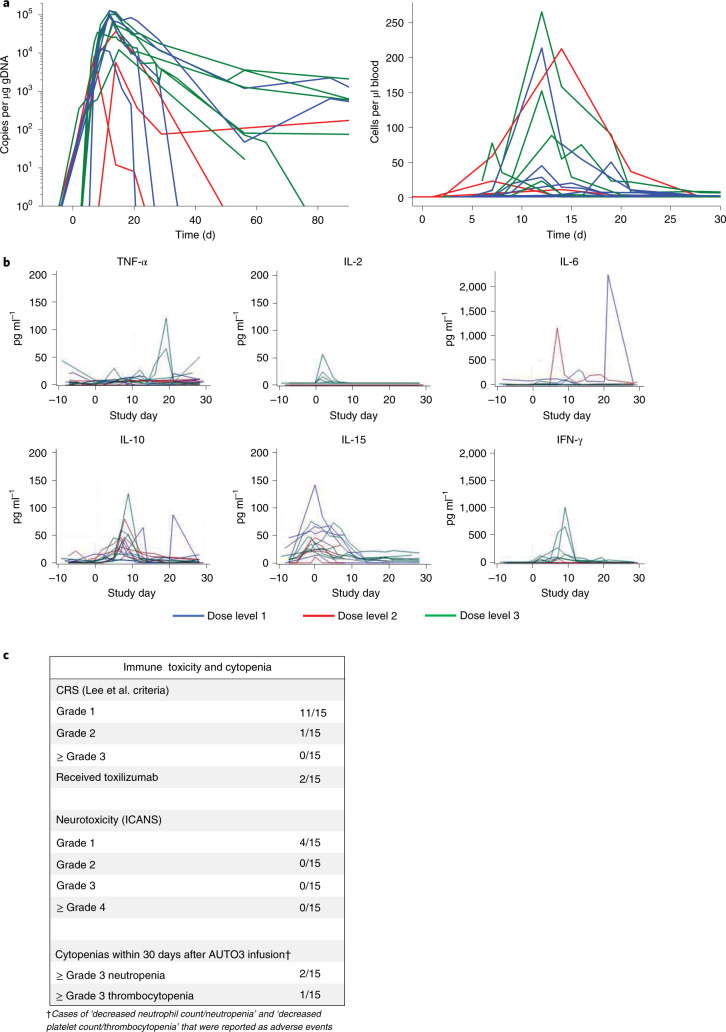
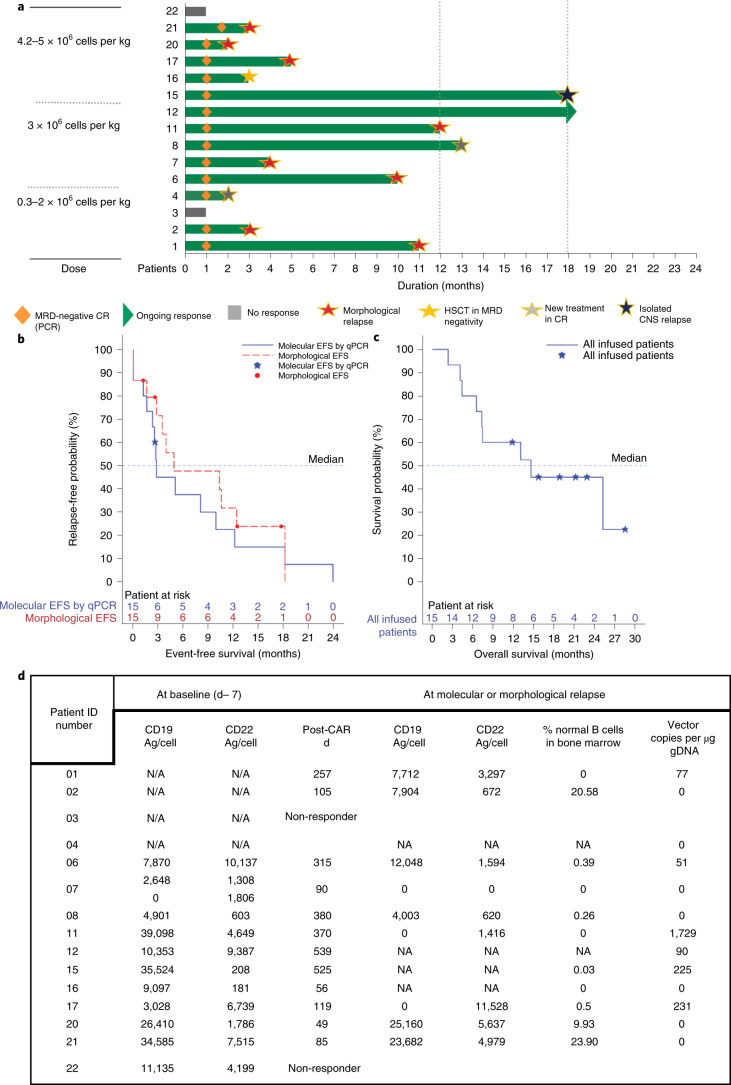
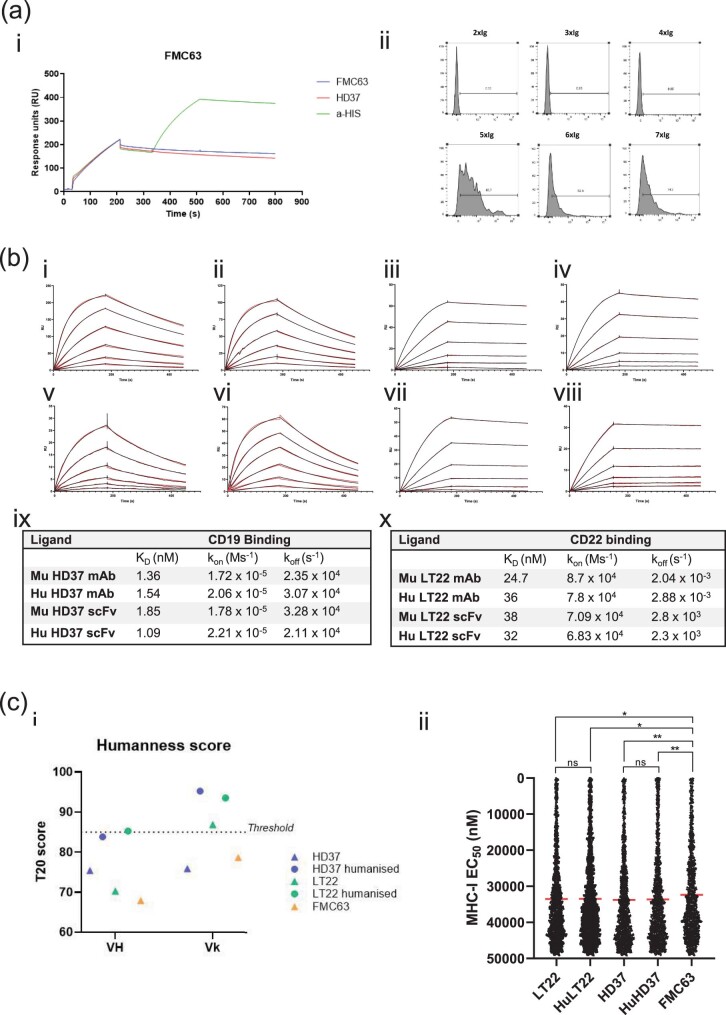
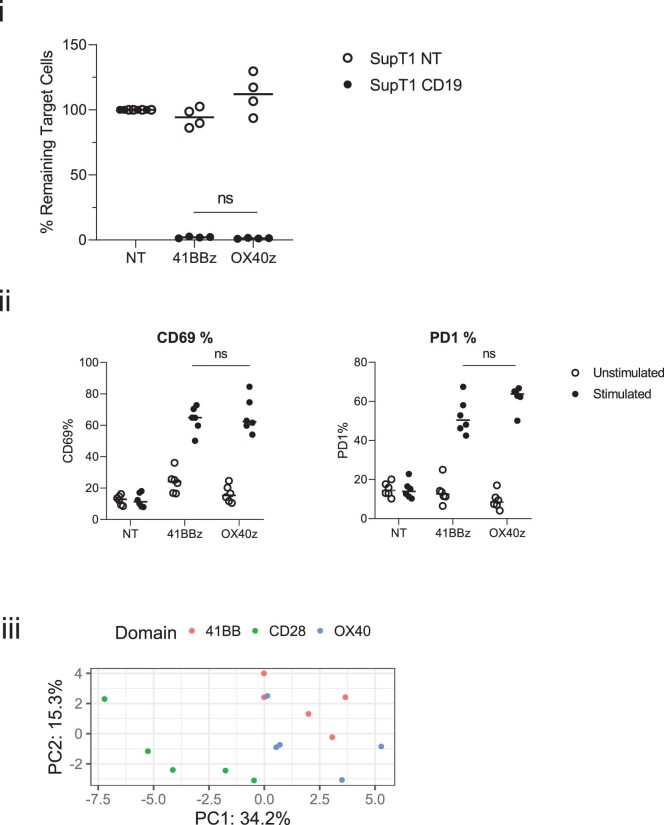
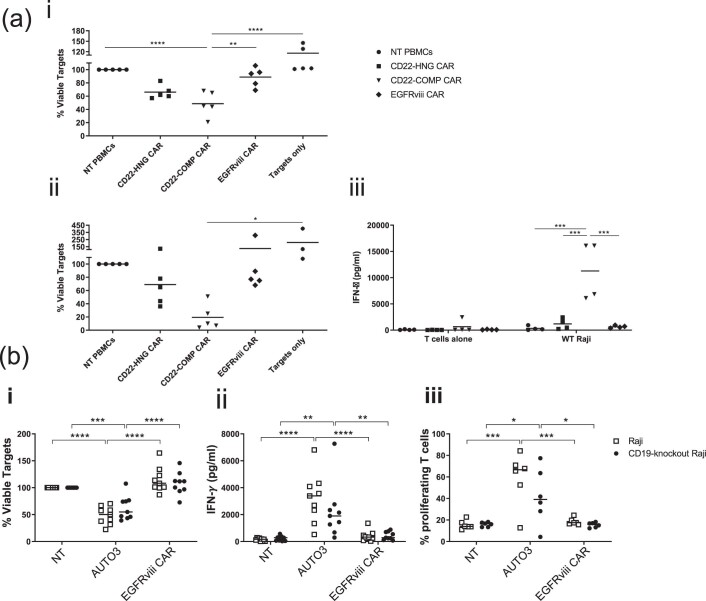
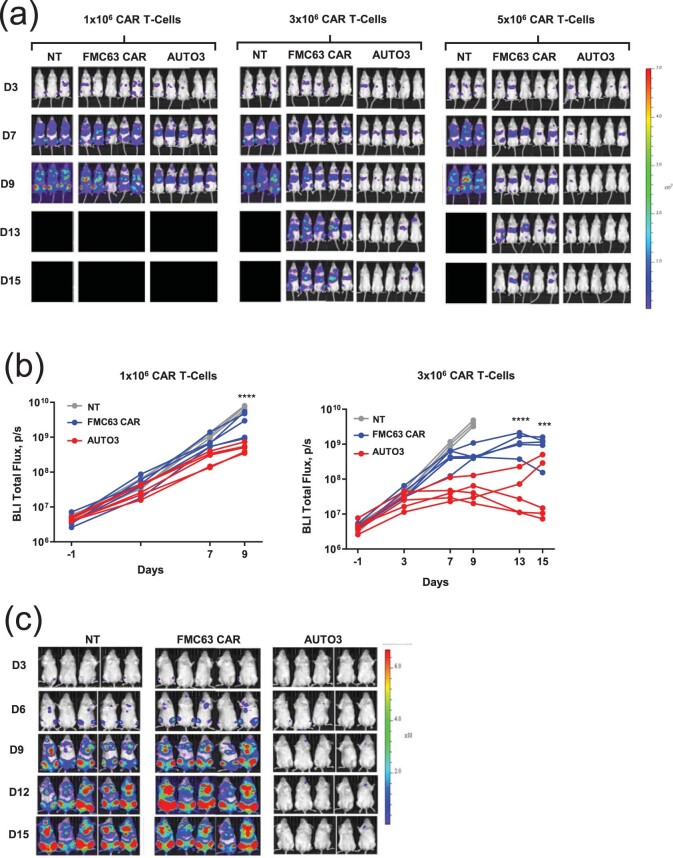
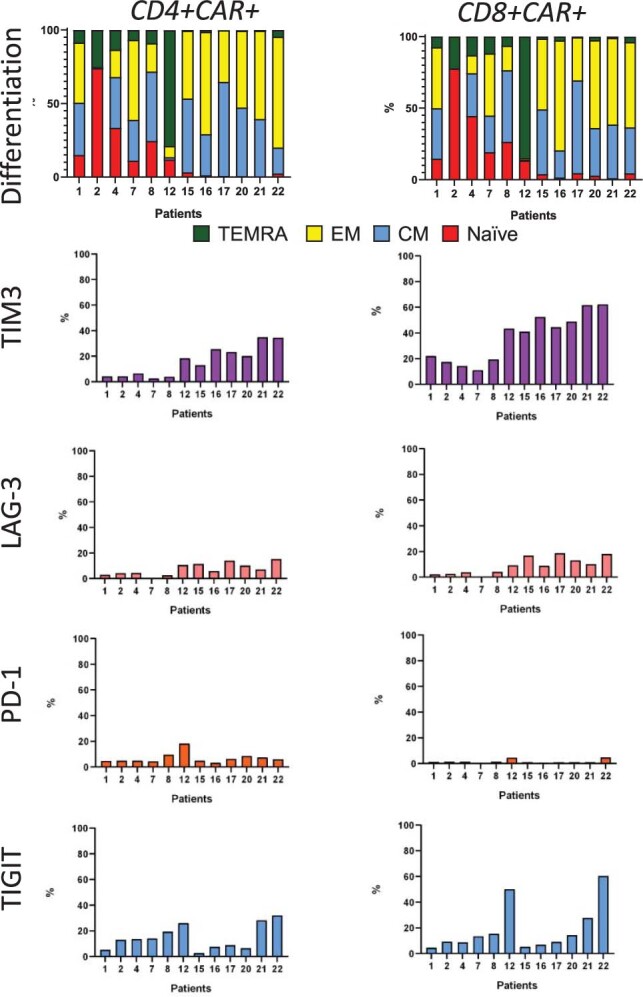
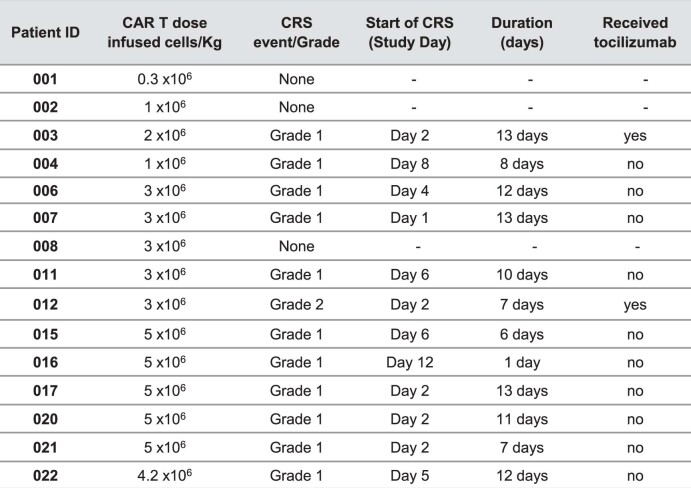
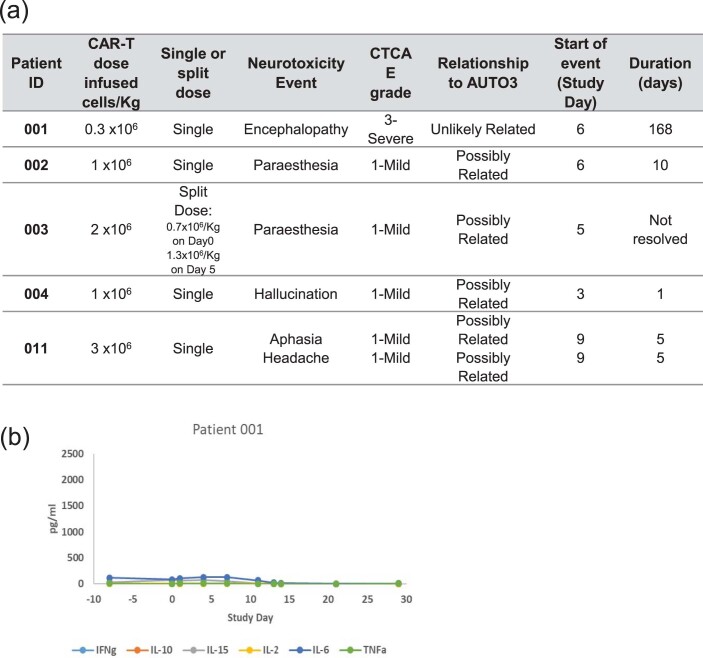
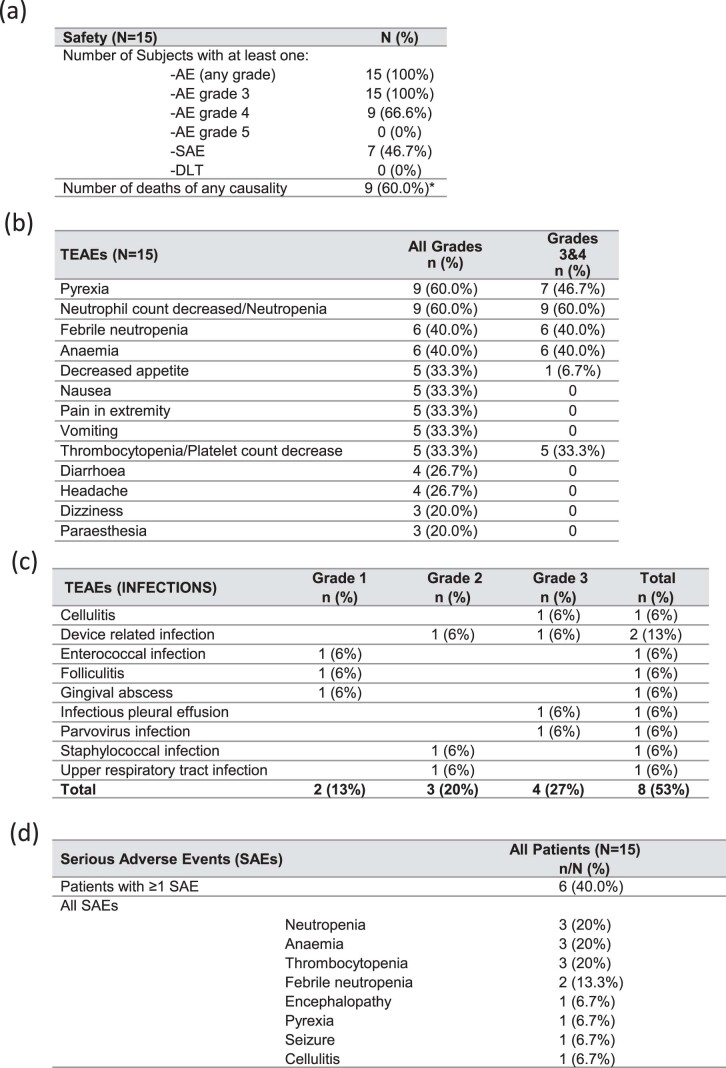
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
