

Aortic root dilatation and dilated cardiomyopathy in an adult with Tatton-Brown-Rahman syndrome
- PMID: 34644003
- PMCID: PMC9175539
- DOI: 10.1002/ajmg.a.62541
Aortic root dilatation and dilated cardiomyopathy in an adult with Tatton-Brown-Rahman syndrome
Abstract
Tatton-Brown-Rahman syndrome is an autosomal dominant overgrowth syndrome caused by pathogenic DNMT3A variants in the germline. Clinical findings of tall stature due to postnatal overgrowth, intellectual disability, and characteristic facial features, are the most consistent findings observed in patients with Tatton-Brown-Rahman syndrome (TBRS). Since the syndrome was first described in 2014, an expanding spectrum of neuropsychiatric, musculoskeletal, neurological, and cardiovascular manifestations have been reported. However, most TBRS cases described in the literature are children with de novo DNMT3A variants, signaling a need to better characterize the phenotypes in adults. In this report, we describe a 34 year old referred to genetics for possible Marfan syndrome with aortic root dilatation, mitral valve prolapse, and dilated cardiomyopathy, who was diagnosed with TBRS due to a heterozygous de novo DNMT3A variant. This represents the third reported TBRS case with aortic root dilation and the second with cardiomyopathy. Collectively, these data provide evidence for an association with aortic disease and cardiomyopathy, highlight the clinical overlap with Marfan syndrome, and suggest that cardiovascular surveillance into adulthood is indicated.
Keywords: DNMT3A; Tatton-Brown-Rahman syndrome; aortic aneurysm; cardiomyopathy; overgrowth syndrome.
© 2021 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Figures
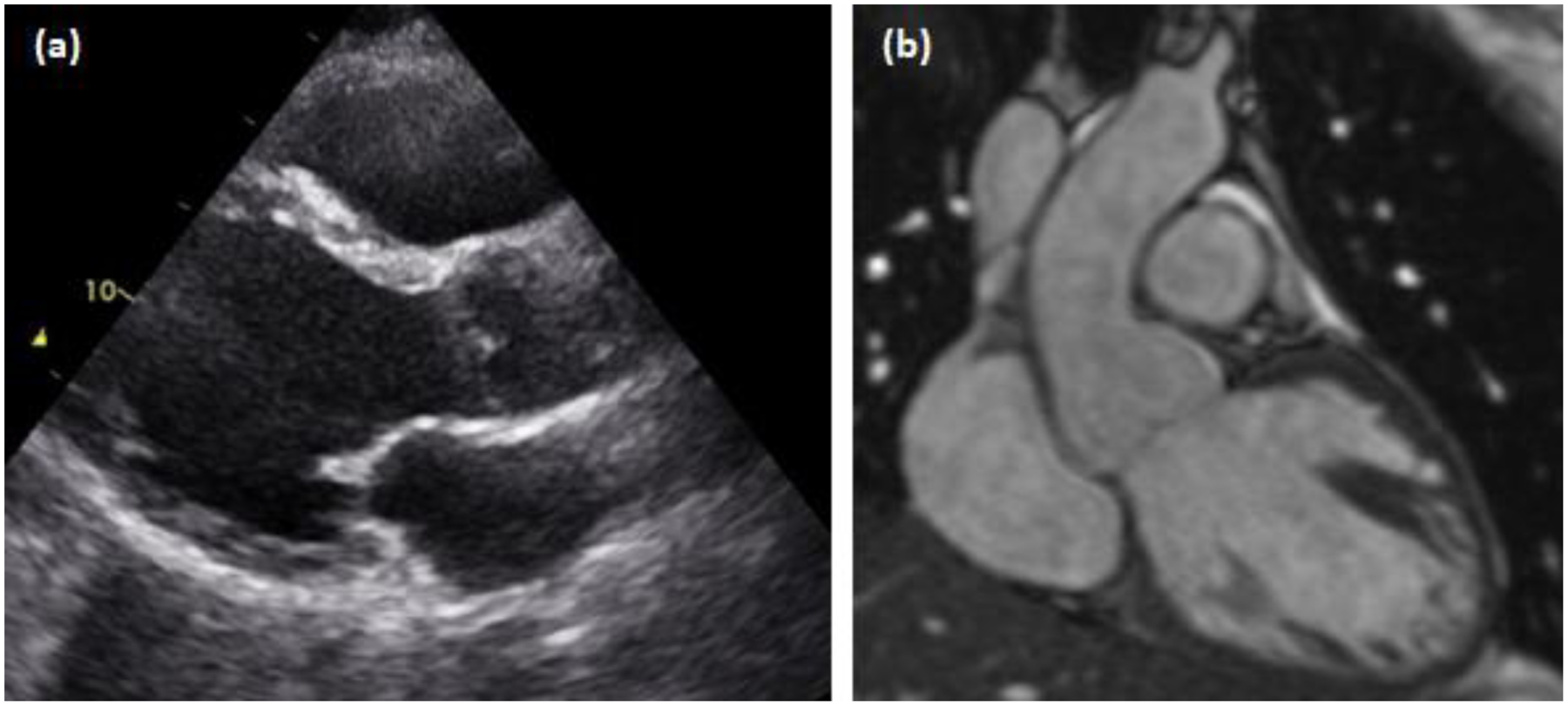
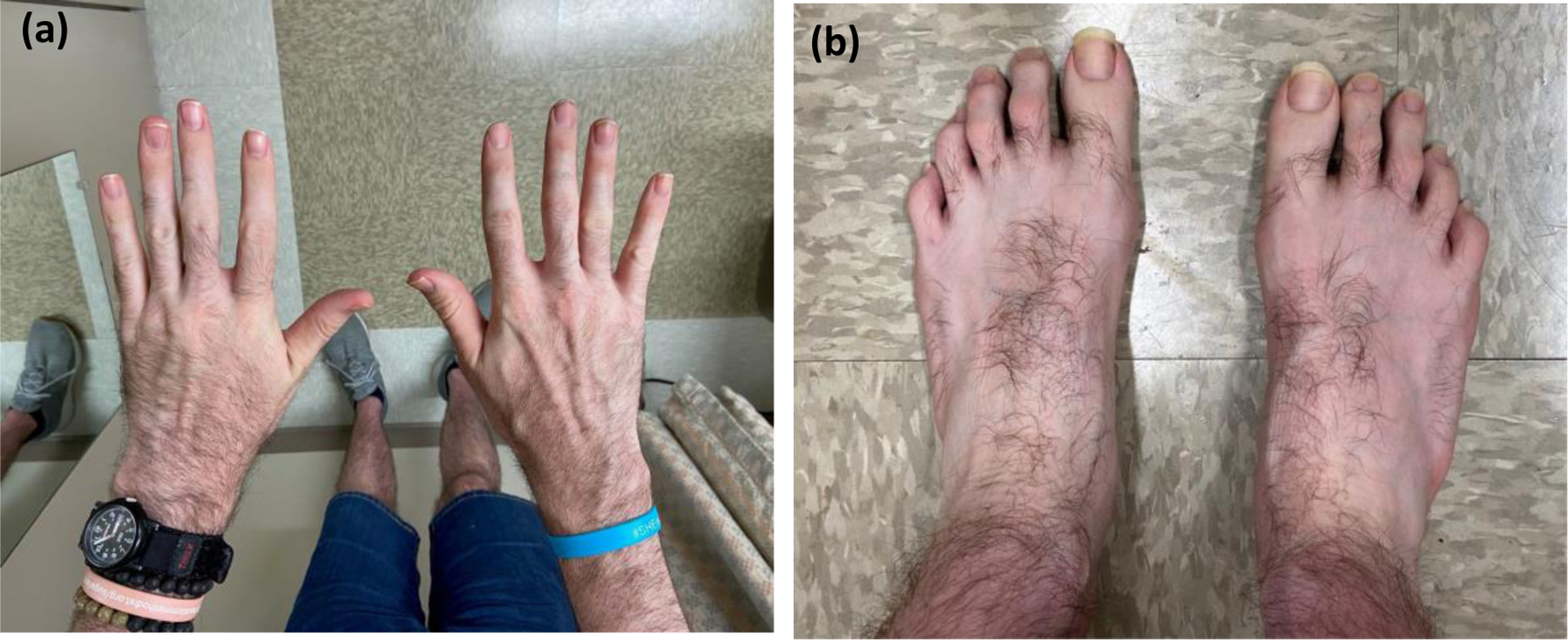
References
-
- Campens L, Demulier L, De Groote K, Vandekerckhove K, De Wolf D, Roman MJ, … De Backer J (2014). Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. The American Journal of Cardiology, 114(6), 914–920. doi:10.1016/j.amjcard.2014.06.024 - DOI - PubMed
-
- Carlston CM, O’Donnell-Luria AH, Underhill HR, Cummings BB, Weisburd B, Minikel EV, … Exome Aggregation Consortium. (2017). Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Human Mutation, 38, 517–523. doi:10.1002/humu.23203 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
