

Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases
- PMID: 34645491
- PMCID: PMC8515723
- DOI: 10.1186/s13073-021-00965-0
Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases
Abstract
Background: Clinical interpretation of genetic variants in the context of the patient's phenotype is becoming the largest component of cost and time expenditure for genome-based diagnosis of rare genetic diseases. Artificial intelligence (AI) holds promise to greatly simplify and speed genome interpretation by integrating predictive methods with the growing knowledge of genetic disease. Here we assess the diagnostic performance of Fabric GEM, a new, AI-based, clinical decision support tool for expediting genome interpretation.
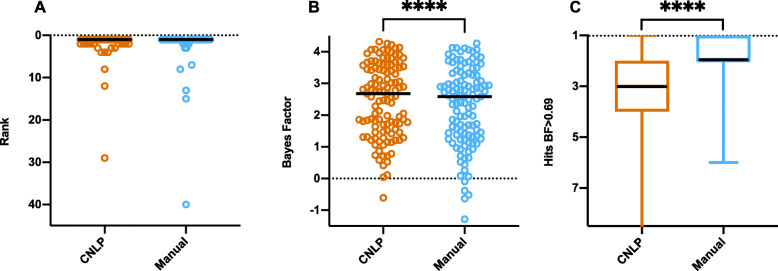
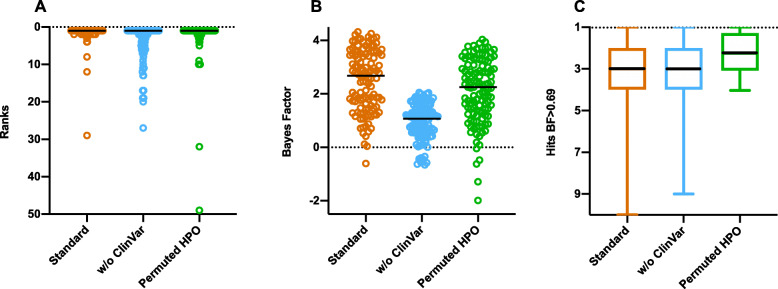
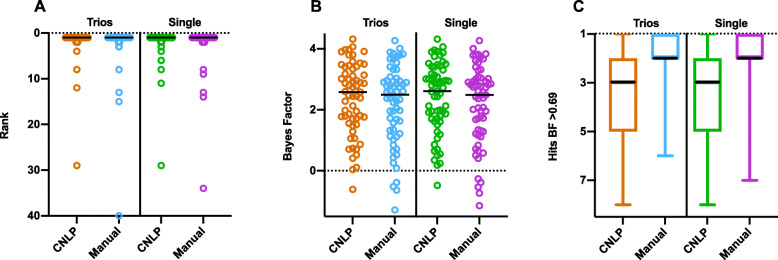
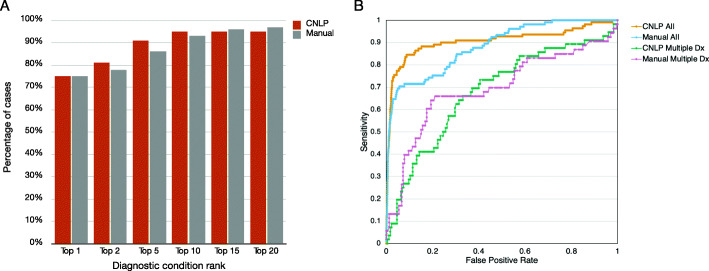
Methods: We benchmarked GEM in a retrospective cohort of 119 probands, mostly NICU infants, diagnosed with rare genetic diseases, who received whole-genome or whole-exome sequencing (WGS, WES). We replicated our analyses in a separate cohort of 60 cases collected from five academic medical centers. For comparison, we also analyzed these cases with current state-of-the-art variant prioritization tools. Included in the comparisons were trio, duo, and singleton cases. Variants underpinning diagnoses spanned diverse modes of inheritance and types, including structural variants (SVs). Patient phenotypes were extracted from clinical notes by two means: manually and using an automated clinical natural language processing (CNLP) tool. Finally, 14 previously unsolved cases were reanalyzed.
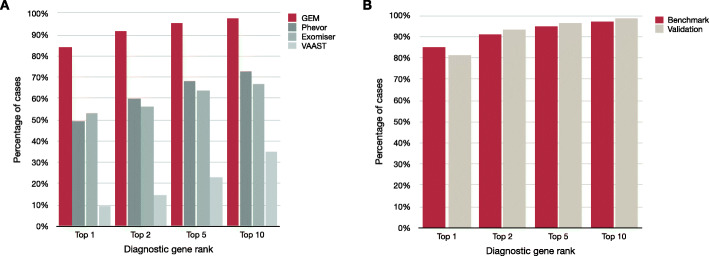
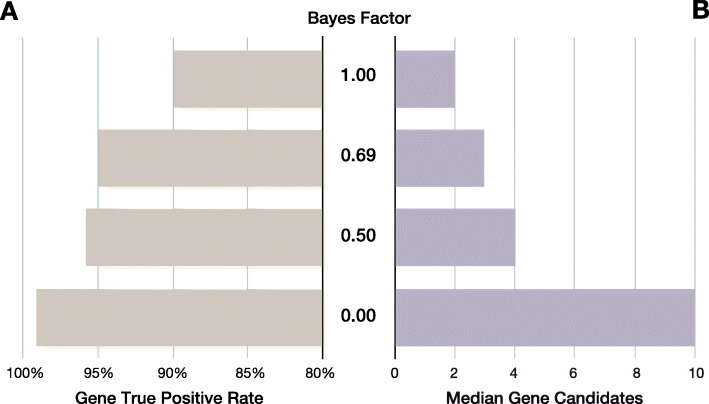
Results: GEM ranked over 90% of the causal genes among the top or second candidate and prioritized for review a median of 3 candidate genes per case, using either manually curated or CNLP-derived phenotype descriptions. Ranking of trios and duos was unchanged when analyzed as singletons. In 17 of 20 cases with diagnostic SVs, GEM identified the causal SVs as the top candidate and in 19/20 within the top five, irrespective of whether SV calls were provided or inferred ab initio by GEM using its own internal SV detection algorithm. GEM showed similar performance in absence of parental genotypes. Analysis of 14 previously unsolved cases resulted in a novel finding for one case, candidates ultimately not advanced upon manual review for 3 cases, and no new findings for 10 cases.
Conclusions: GEM enabled diagnostic interpretation inclusive of all variant types through automated nomination of a very short list of candidate genes and disorders for final review and reporting. In combination with deep phenotyping by CNLP, GEM enables substantial automation of genetic disease diagnosis, potentially decreasing cost and expediting case review.
© 2021. The Author(s).
Conflict of interest statement
FV, EF, JM, and MGR were employees of Fabric Genomics Inc. during the performance of this work and have received stock grants from Fabric Genomics Inc. BM, PB, and MY are consultants to Fabric Genomics Inc. and have received consulting fees and stock grants from Fabric Genomics Inc. The remaining authors declare that they have no competing interests.
Figures
References
-
- Online Mendelian Inheritance in Man, OMIM®McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) (available at https://omim.org/).
-
- Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. 2018;10:1–16. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
