

Systems-level effects of allosteric perturbations to a model molecular switch
- PMID: 34646016
- PMCID: PMC8571063
- DOI: 10.1038/s41586-021-03982-6
Systems-level effects of allosteric perturbations to a model molecular switch
Abstract
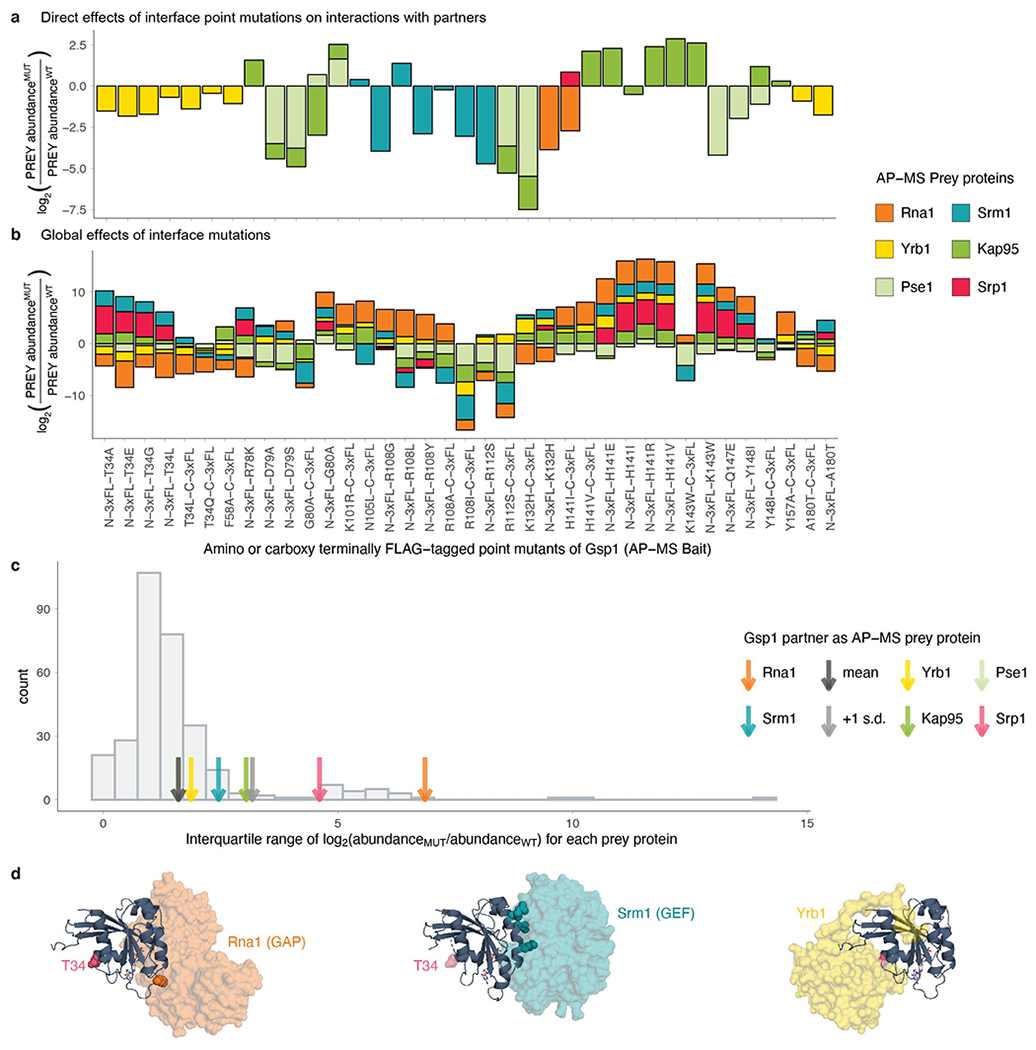
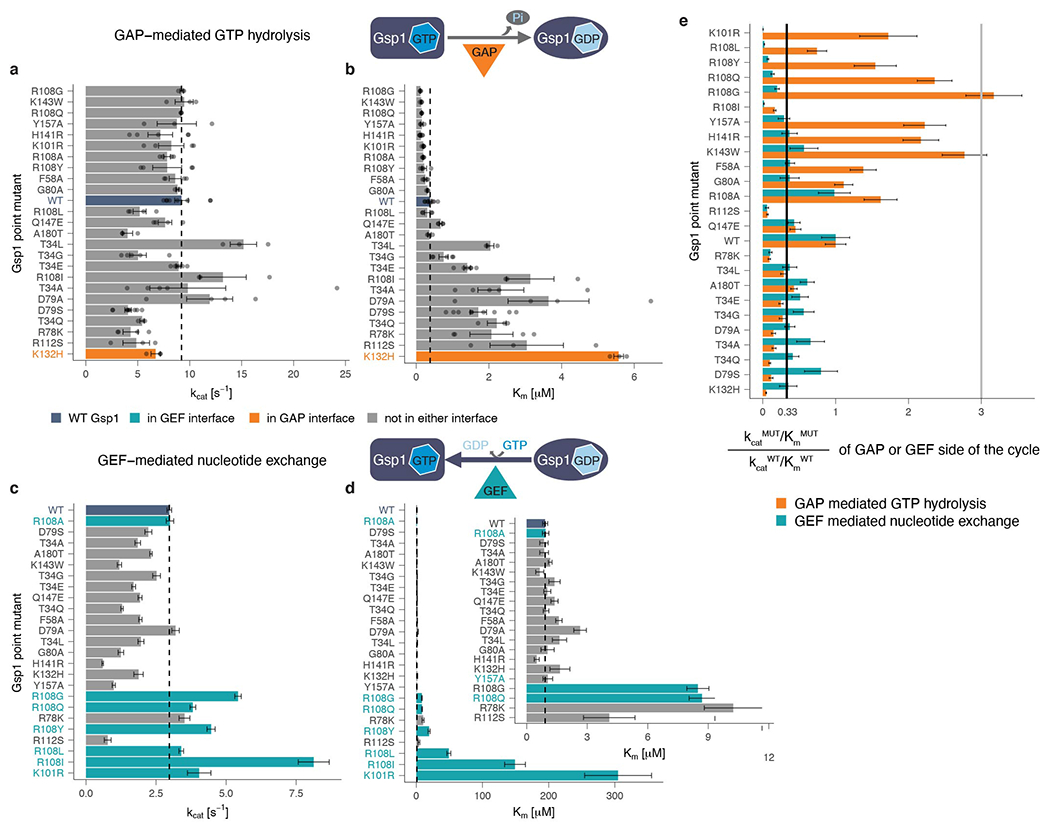
Molecular switch proteins whose cycling between states is controlled by opposing regulators1,2 are central to biological signal transduction. As switch proteins function within highly connected interaction networks3, the fundamental question arises of how functional specificity is achieved when different processes share common regulators. Here we show that functional specificity of the small GTPase switch protein Gsp1 in Saccharomyces cerevisiae (the homologue of the human protein RAN)4 is linked to differential sensitivity of biological processes to different kinetics of the Gsp1 (RAN) switch cycle. We make 55 targeted point mutations to individual protein interaction interfaces of Gsp1 (RAN) and show through quantitative genetic5 and physical interaction mapping that Gsp1 (RAN) interface perturbations have widespread cellular consequences. Contrary to expectation, the cellular effects of the interface mutations group by their biophysical effects on kinetic parameters of the GTPase switch cycle and not by the targeted interfaces. Instead, we show that interface mutations allosterically tune the GTPase cycle kinetics. These results suggest a model in which protein partner binding, or post-translational modifications at distal sites, could act as allosteric regulators of GTPase switching. Similar mechanisms may underlie regulation by other GTPases, and other biological switches. Furthermore, our integrative platform to determine the quantitative consequences of molecular perturbations may help to explain the effects of disease mutations that target central molecular switches.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures












Comment in
-
A kinetic mechanism for systems-level behavior in GTPase signaling.Trends Biochem Sci. 2022 Jun;47(6):459-460. doi: 10.1016/j.tibs.2022.01.006. Epub 2022 Feb 19. Trends Biochem Sci. 2022. PMID: 35190221 Free PMC article.
References
Main Text References
Additional References Cited in Main Methods
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
