

Tissue factor cytoplasmic domain exacerbates post-infarct left ventricular remodeling via orchestrating cardiac inflammation and angiogenesis
- PMID: 34646369
- PMCID: PMC8490508
- DOI: 10.7150/thno.63354
Tissue factor cytoplasmic domain exacerbates post-infarct left ventricular remodeling via orchestrating cardiac inflammation and angiogenesis
Abstract
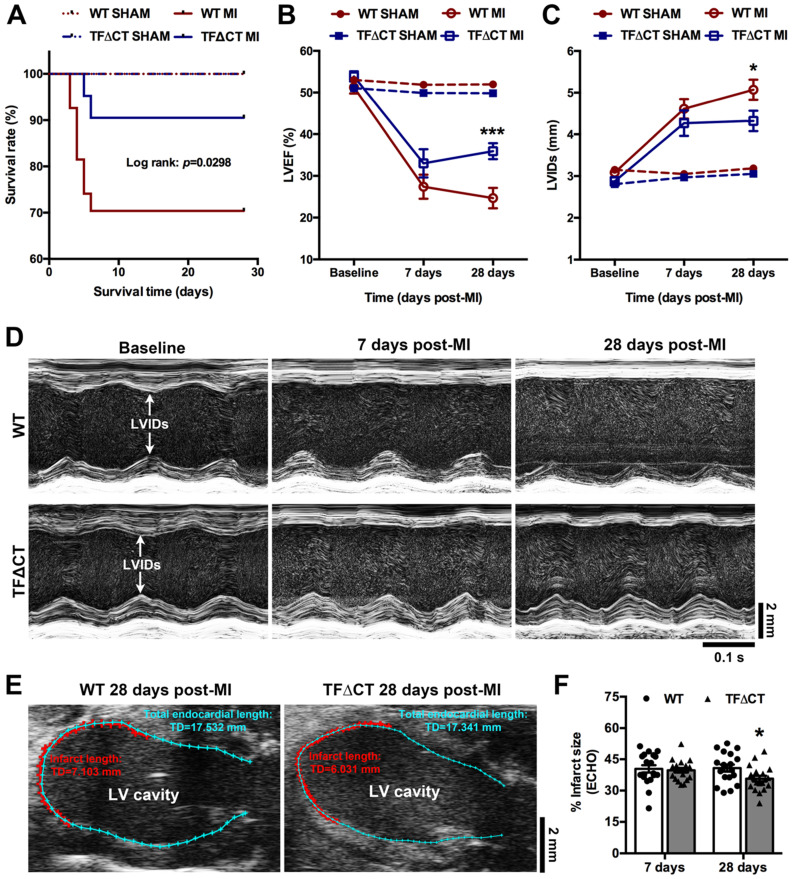
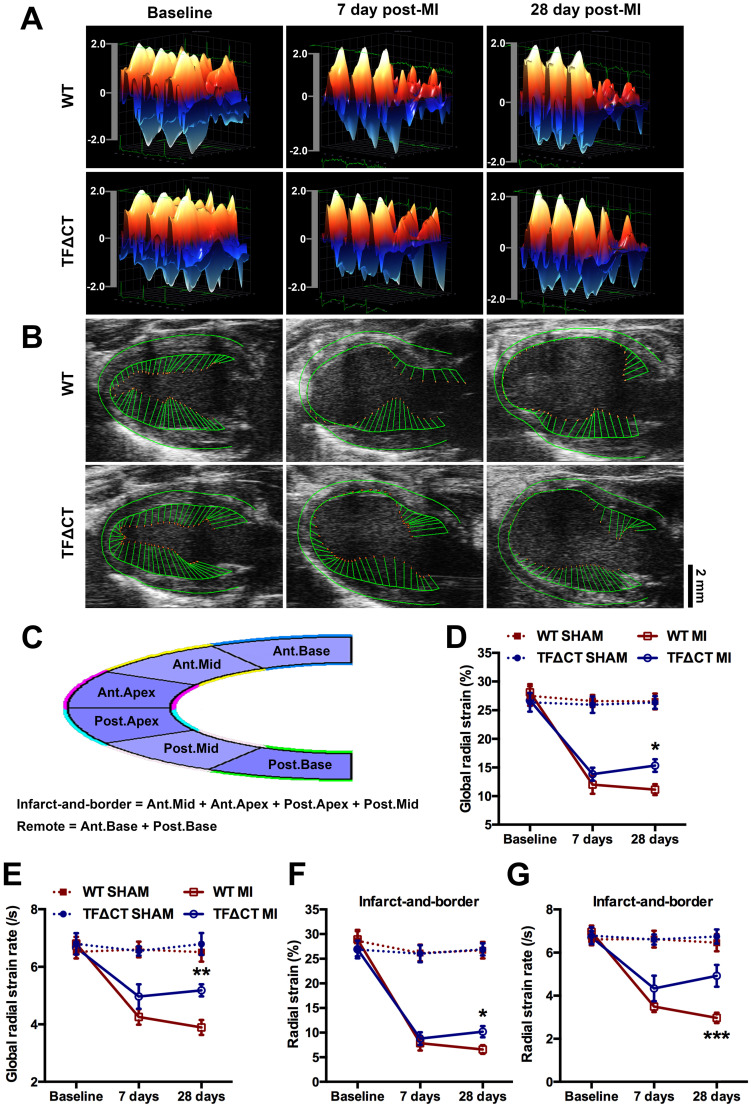
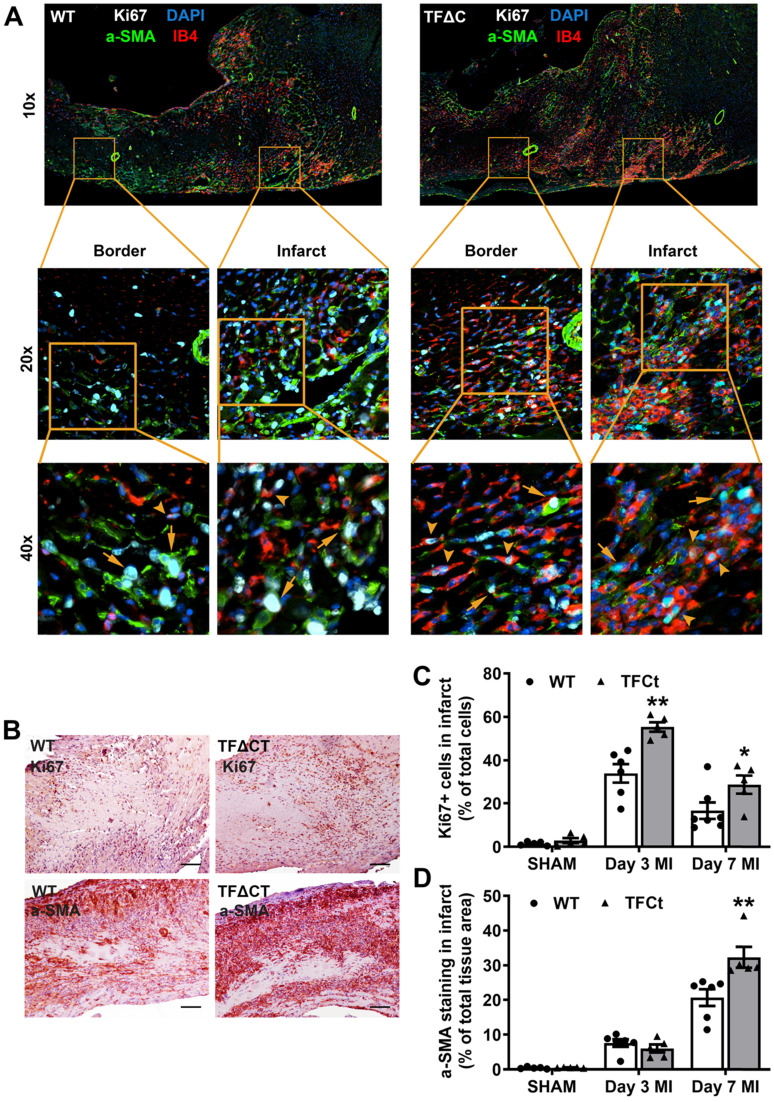
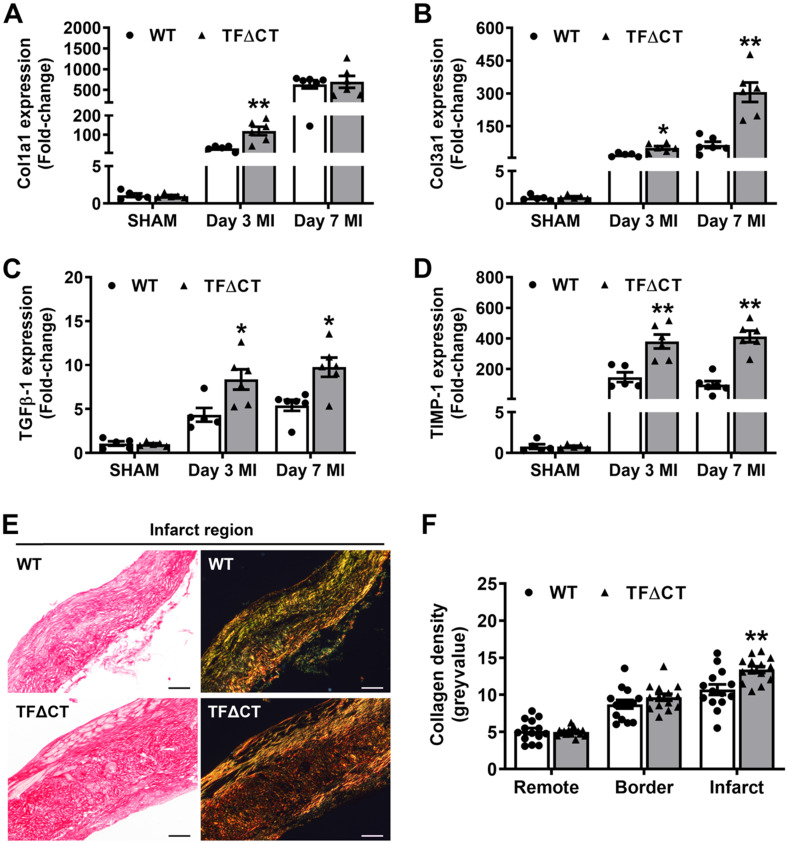
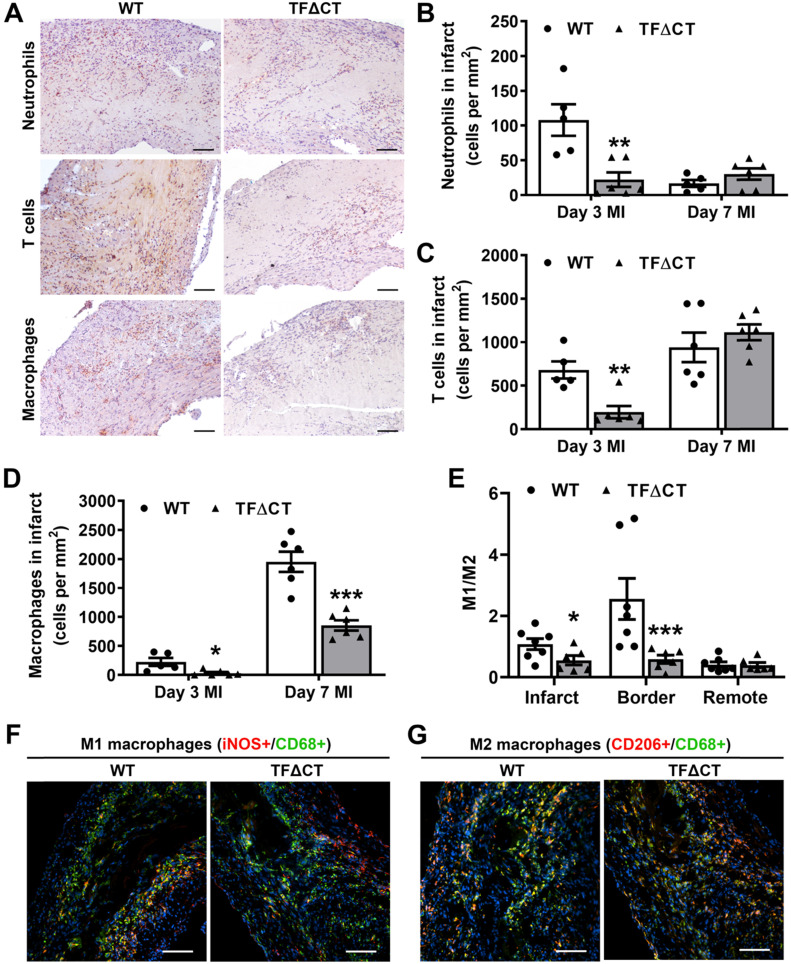
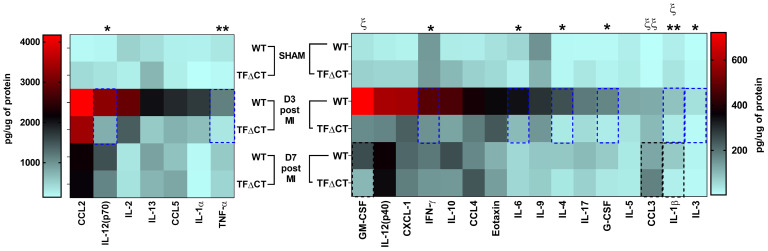
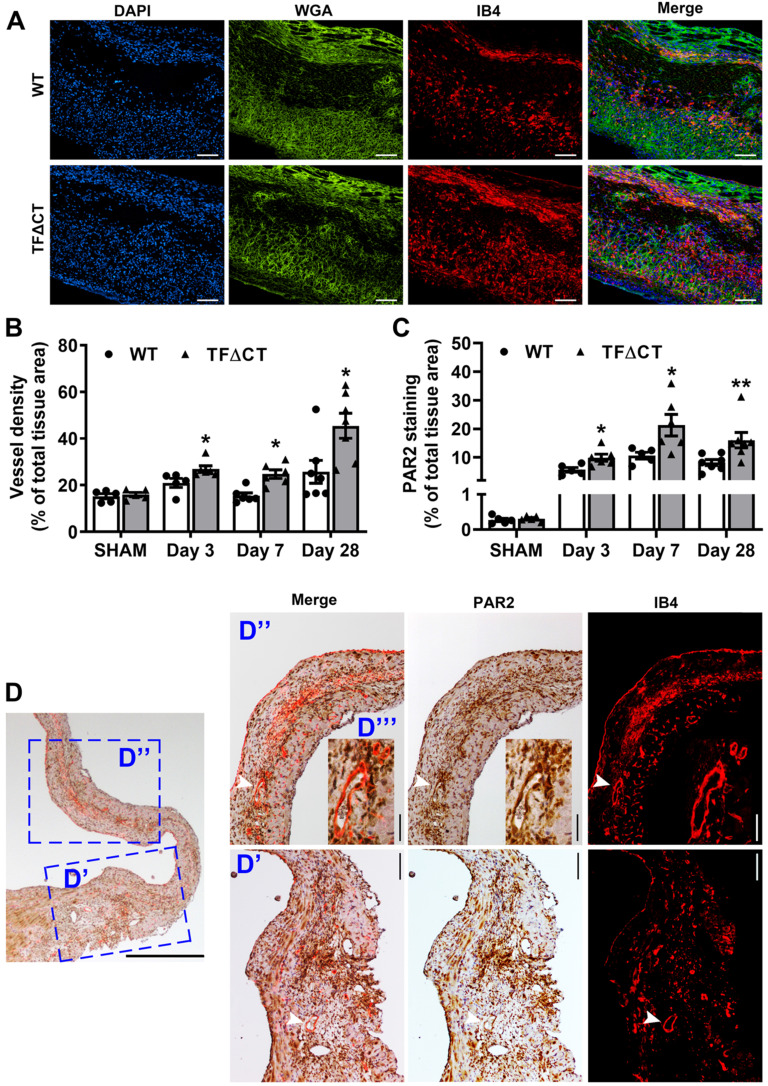
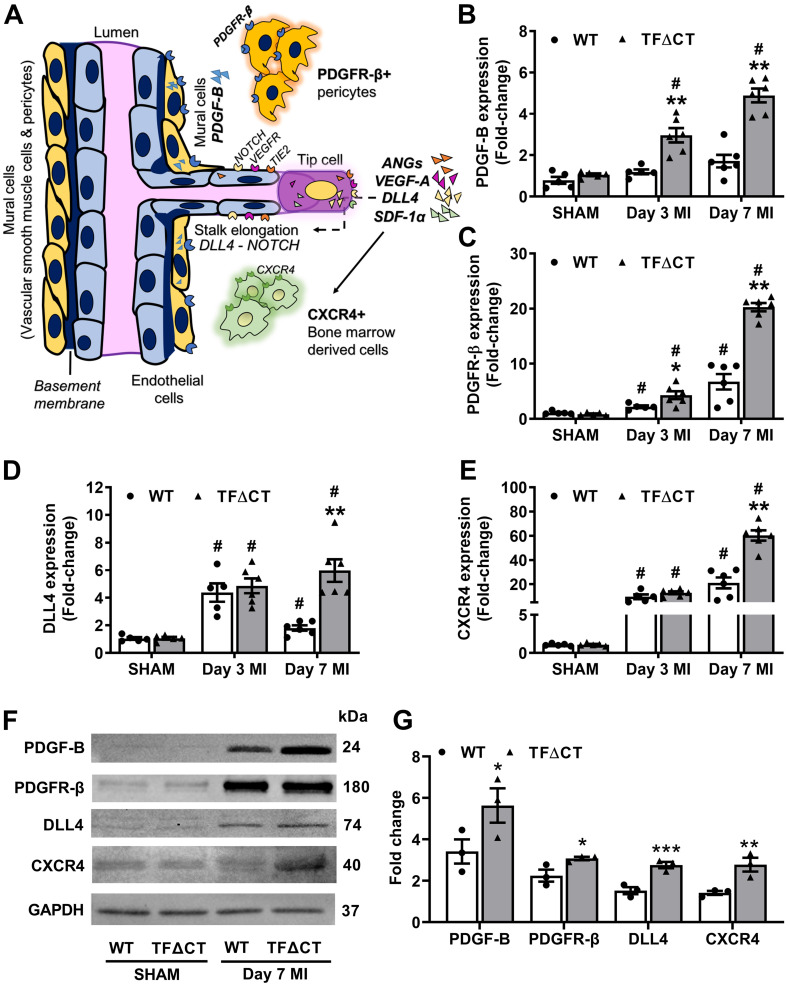
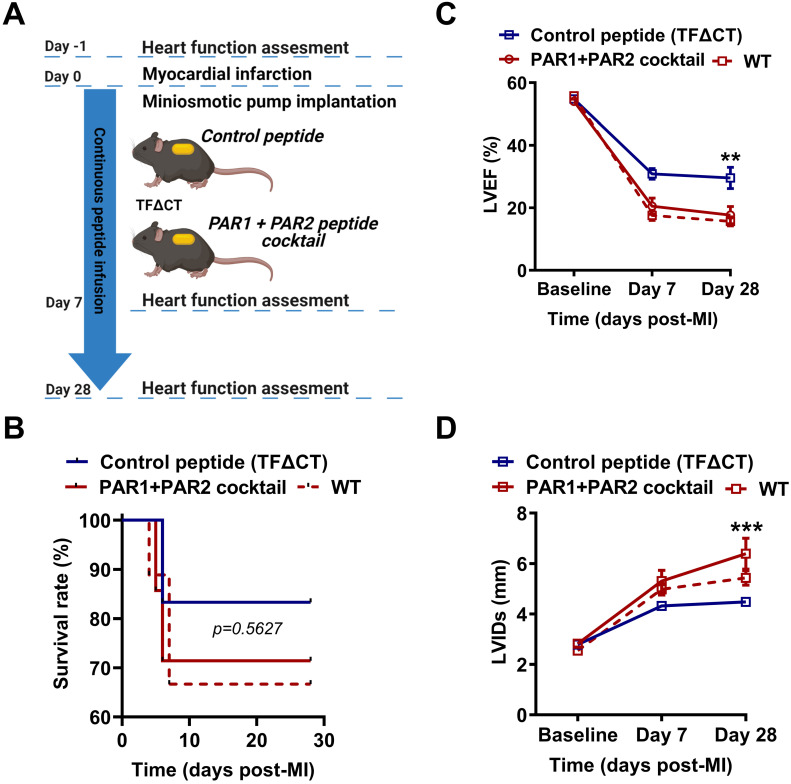
The coagulation protein tissue factor (TF) regulates inflammation and angiogenesis via its cytoplasmic domain in infection, cancer and diabetes. While TF is highly abundant in the heart and is implicated in cardiac pathology, the contribution of its cytoplasmic domain to post-infarct myocardial injury and adverse left ventricular (LV) remodeling remains unknown. Methods: Myocardial infarction was induced in wild-type mice or mice lacking the TF cytoplasmic domain (TF∆CT) by occlusion of the left anterior descending coronary artery. Heart function was monitored with echocardiography. Heart tissue was collected at different time-points for histological, molecular and flow cytometry analysis. Results: Compared with wild-type mice, TF∆CT had a higher survival rate during a 28-day follow-up after myocardial infarction. Among surviving mice, TF∆CT mice had better cardiac function and less LV remodeling than wild-type mice. The overall improvement of post-infarct cardiac performance in TF∆CT mice, as revealed by speckle-tracking strain analysis, was attributed to reduced myocardial deformation in the peri-infarct region. Histological analysis demonstrated that TF∆CT hearts had in the infarct area greater proliferation of myofibroblasts and better scar formation. Compared with wild-type hearts, infarcted TF∆CT hearts showed less infiltration of proinflammatory cells with concomitant lower expression of protease-activated receptor-1 (PAR1) - Rac1 axis. In particular, infarcted TF∆CT hearts displayed markedly lower ratios of inflammatory M1 macrophages and reparative M2 macrophages (M1/M2). In vitro experiment with primary macrophages demonstrated that deletion of the TF cytoplasmic domain inhibited macrophage polarization toward the M1 phenotype. Furthermore, infarcted TF∆CT hearts presented markedly higher peri-infarct vessel density associated with enhanced endothelial cell proliferation and higher expression of PAR2 and PAR2-associated pro-angiogenic pathway factors. Finally, the overall cardioprotective effects observed in TF∆CT mice could be abolished by subcutaneously infusing a cocktail of PAR1-activating peptide and PAR2-inhibiting peptide via osmotic minipumps. Conclusions: Our findings demonstrate that the TF cytoplasmic domain exacerbates post-infarct cardiac injury and adverse LV remodeling via differential regulation of inflammation and angiogenesis. Targeted inhibition of the TF cytoplasmic domain-mediated intracellular signaling may ameliorate post-infarct LV remodeling without perturbing coagulation.
Keywords: adverse left ventricular remodeling; angiogenesis; inflammation; myocardial infarction; tissue factor cytoplasmic domain.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- D'Alessandro E, Posma JJN, Spronk HMH, Ten Cate H. Tissue factor (:Factor VIIa) in the heart and vasculature: More than an envelope. Thromb Res. 2018;168:130–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
