

Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization
- PMID: 34647815
- PMCID: PMC8570347
- DOI: 10.1161/CIRCULATIONAHA.120.052023
Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization
Abstract
Background: Long noncoding RNAs (lncRNAs) are important regulators of biological processes involved in vascular tissue homeostasis and disease development. The present study assessed the functional contribution of the lncRNA myocardial infarction-associated transcript (MIAT) to atherosclerosis and carotid artery disease.
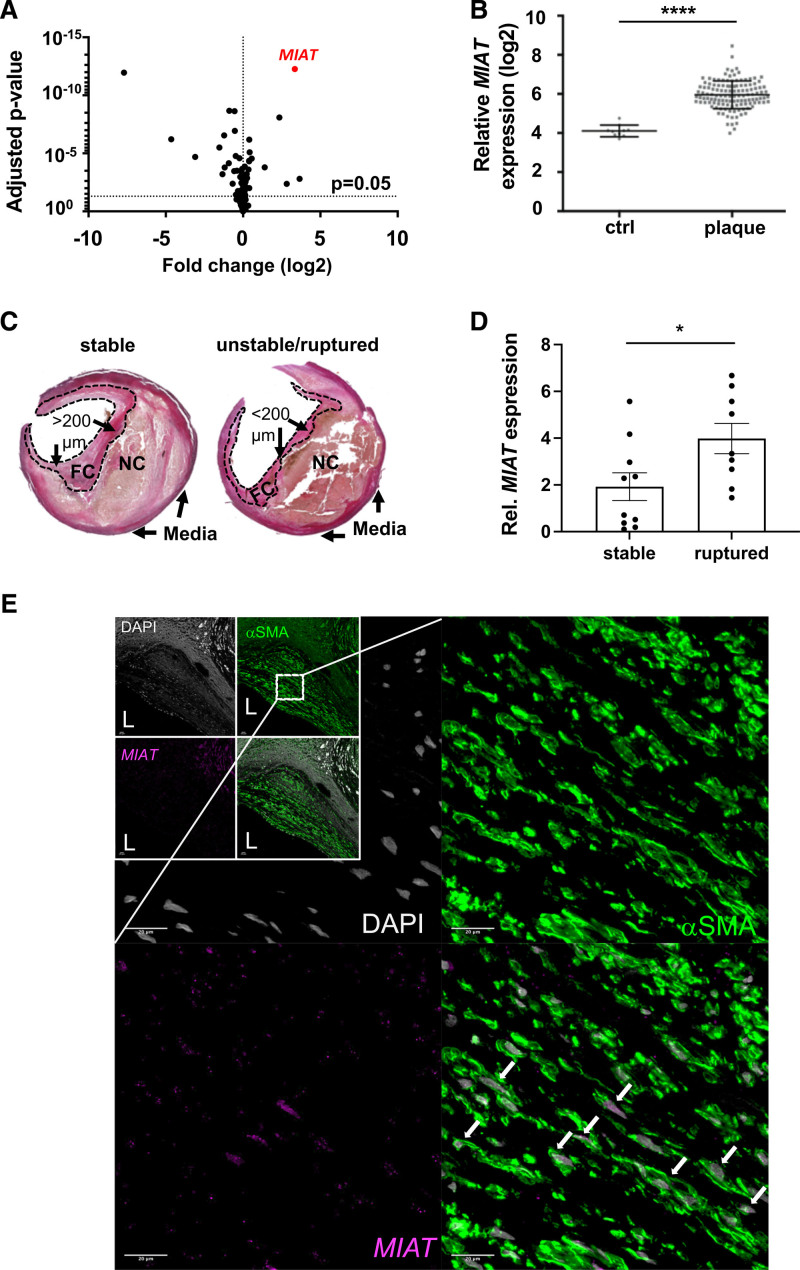
Methods: We profiled differences in RNA transcript expression in patients with advanced carotid artery atherosclerotic lesions from the Biobank of Karolinska Endarterectomies. The lncRNA MIAT was identified as the most upregulated noncoding RNA transcript in carotid plaques compared with nonatherosclerotic control arteries, which was confirmed by quantitative real-time polymerase chain reaction and in situ hybridization.
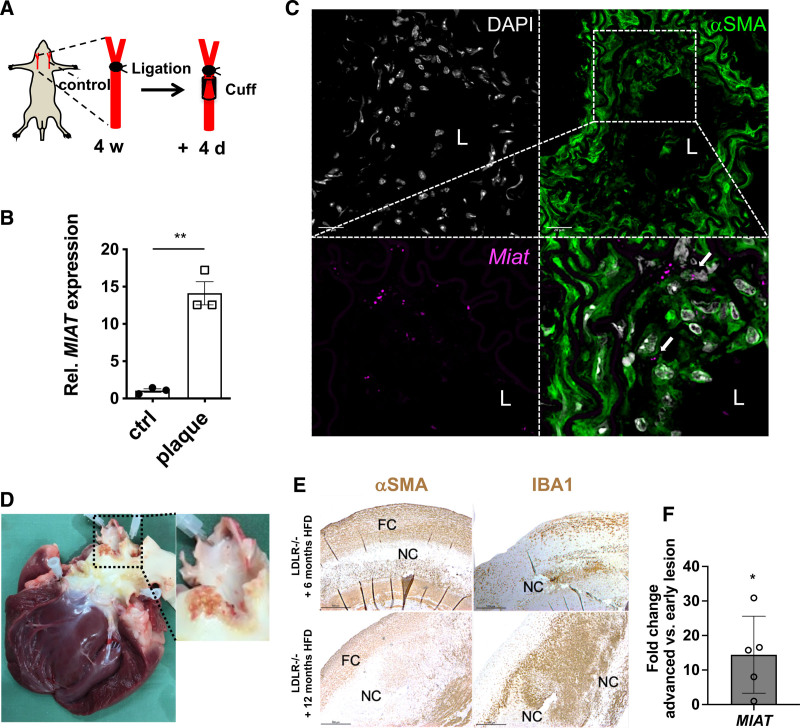
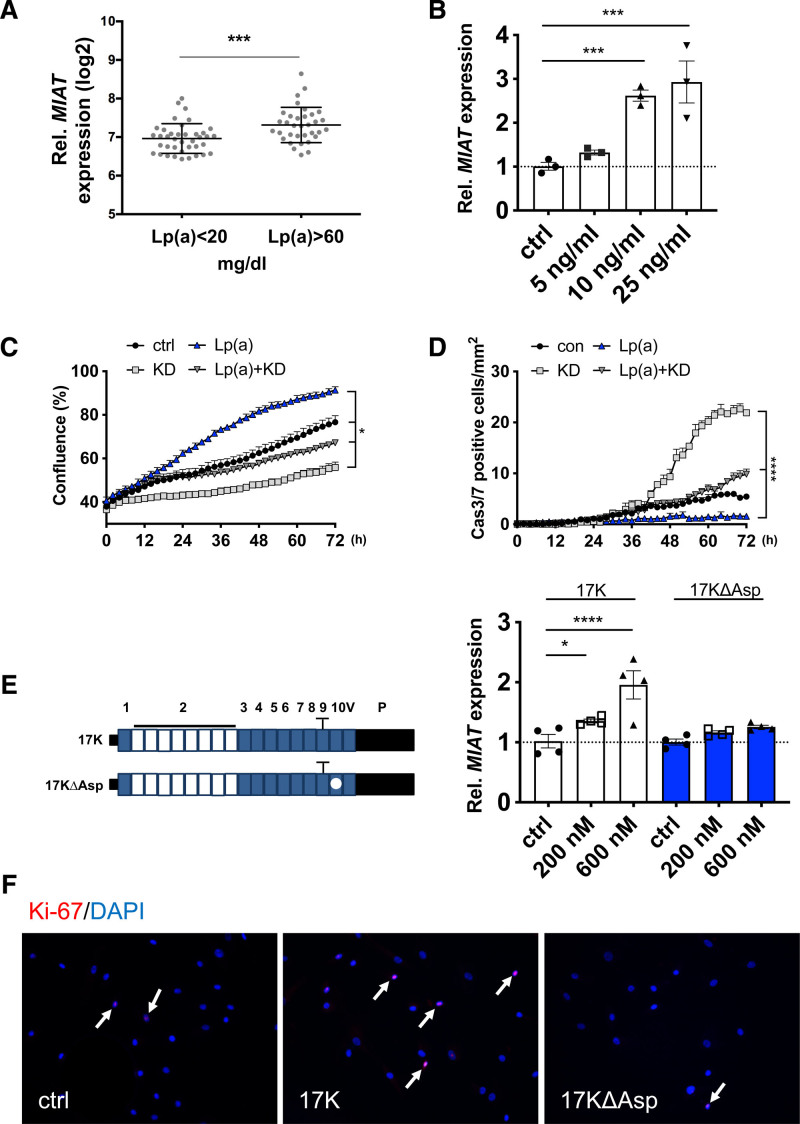
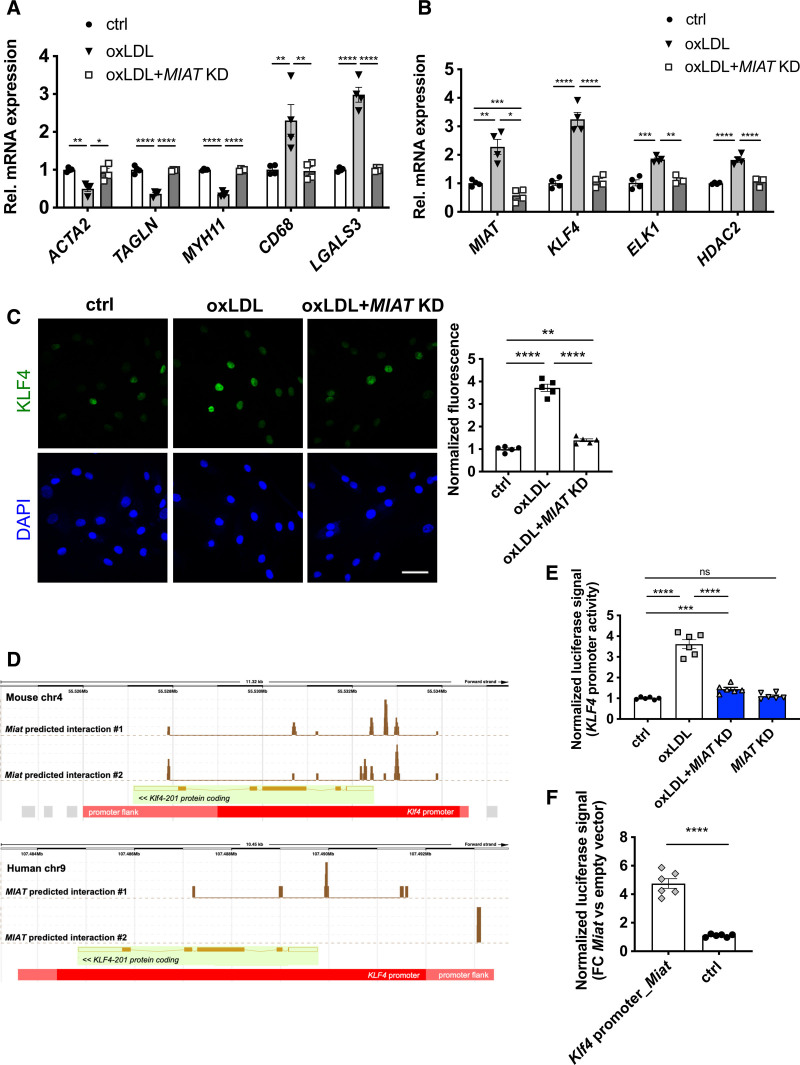
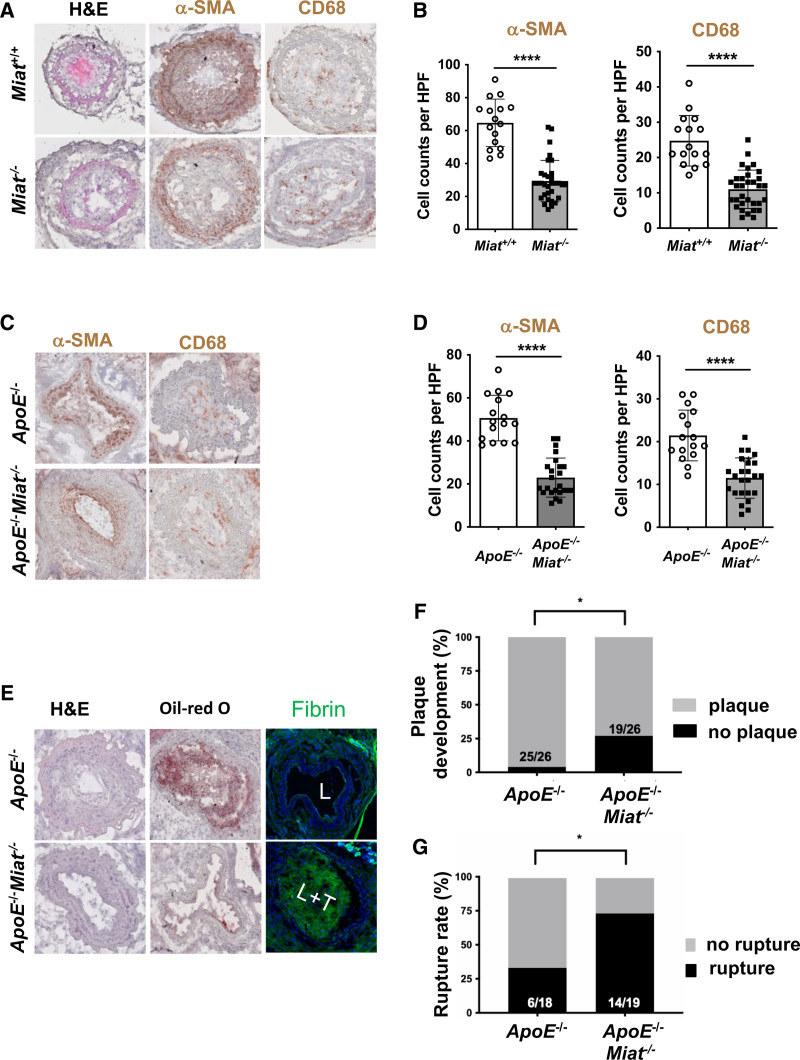
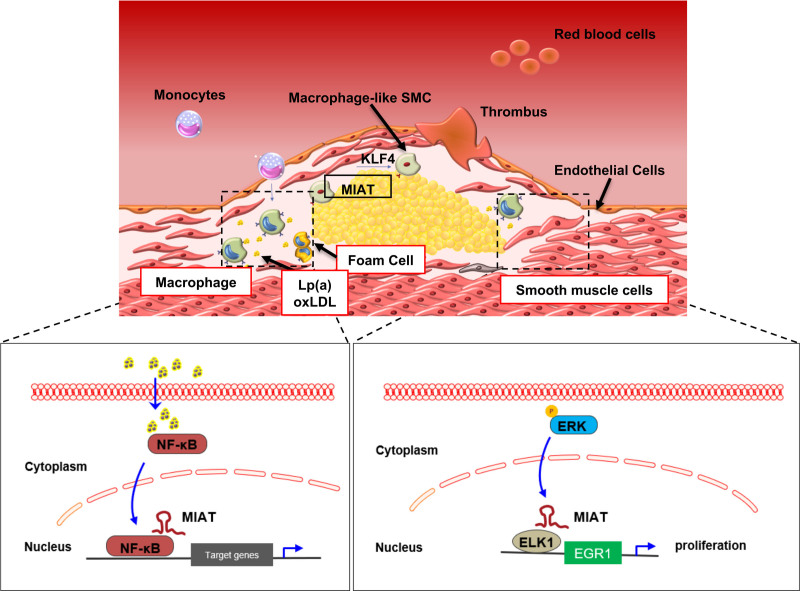
Results: Experimental knockdown of MIAT, using site-specific antisense oligonucleotides (LNA-GapmeRs) not only markedly decreased proliferation and migration rates of cultured human carotid artery smooth muscle cells (SMCs) but also increased their apoptosis. MIAT mechanistically regulated SMC proliferation through the EGR1 (Early Growth Response 1)-ELK1 (ETS Transcription Factor ELK1)-ERK (Extracellular Signal-Regulated Kinase) pathway. MIAT is further involved in SMC phenotypic transition to proinflammatory macrophage-like cells through binding to the promoter region of KLF4 and enhancing its transcription. Studies using Miat-/- and Miat-/-ApoE-/- mice, and Yucatan LDLR-/- mini-pigs, as well, confirmed the regulatory role of this lncRNA in SMC de- and transdifferentiation and advanced atherosclerotic lesion formation.
Conclusions: The lncRNA MIAT is a novel regulator of cellular processes in advanced atherosclerosis that controls proliferation, apoptosis, and phenotypic transition of SMCs, and the proinflammatory properties of macrophages, as well.
Keywords: RNA, long noncoding; atherosclerosis; carotid artery diseases; lipoprotein(a); myocytes, smooth muscle; stroke.
Figures


References
-
- van Lammeren GW, den Ruijter HM, Vrijenhoek JE, van der Laan SW, Velema E, de Vries JP, de Kleijn DP, Vink A, de Borst GJ, Moll FL, et al. . Time-dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery. Circulation. 2014; 129:2269–2276. doi: 10.1161/CIRCULATIONAHA.113.007603 - PubMed
-
- Fernández-Friera L, Peñalvo JL, Fernández-Ortiz A, Ibañez B, López-Melgar B, Laclaustra M, Oliva B, Mocoroa A, Mendiguren J, Martínez de Vega V, et al. . Prevalence, vascular distribution, and multiterritorial extent of subclinical atherosclerosis in a middle-aged cohort: the PESA (Progression of Early Subclinical Atherosclerosis) study. Circulation. 2015; 131:2104–2113. doi: 10.1161/CIRCULATIONAHA.114.014310 - PubMed
-
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008; 371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7 - PubMed
-
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011; 473:317–325. doi: 10.1038/nature10146 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
