

Quality control and evaluation of plant epigenomics data
- PMID: 34648025
- PMCID: PMC8773985
- DOI: 10.1093/plcell/koab255
Quality control and evaluation of plant epigenomics data
Abstract
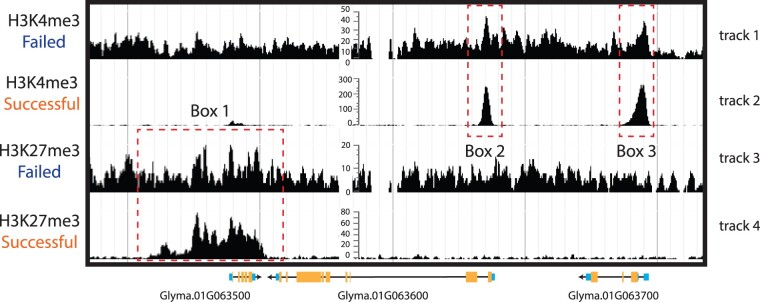
Epigenomics is the study of molecular signatures associated with discrete regions within genomes, many of which are important for a wide range of nuclear processes. The ability to profile the epigenomic landscape associated with genes, repetitive regions, transposons, transcription, differential expression, cis-regulatory elements, and 3D chromatin interactions has vastly improved our understanding of plant genomes. However, many epigenomic and single-cell genomic assays are challenging to perform in plants, leading to a wide range of data quality issues; thus, the data require rigorous evaluation prior to downstream analyses and interpretation. In this commentary, we provide considerations for the evaluation of plant epigenomics and single-cell genomics data quality with the aim of improving the quality and utility of studies using those data across diverse plant species.
© The Author(s) 2021. Published by Oxford University Press on behalf of American Society of Plant Biologists.
Figures

References
-
- Axtell MJ (2013a) Classification and comparison of small RNAs from plants. Annu Rev Plant Biol 64:137–159 - PubMed
-
- Blondel VD, Guilamume J, Lambiotte R, Lefebvre E (2008) Fast unfolding of communities in large networks. J Stat Mech 2008 DOI:10.1088/1742-5468/2008/10/P10008
