

Genomic reconstruction of the SARS-CoV-2 epidemic in England
- PMID: 34649268
- PMCID: PMC8674138
- DOI: 10.1038/s41586-021-04069-y
Genomic reconstruction of the SARS-CoV-2 epidemic in England
Erratum in
-
Publisher Correction: Genomic reconstruction of the SARS CoV-2 epidemic in England.Nature. 2022 Jun;606(7915):E18. doi: 10.1038/s41586-022-04887-8. Nature. 2022. PMID: 35701578 Free PMC article. No abstract available.
Abstract
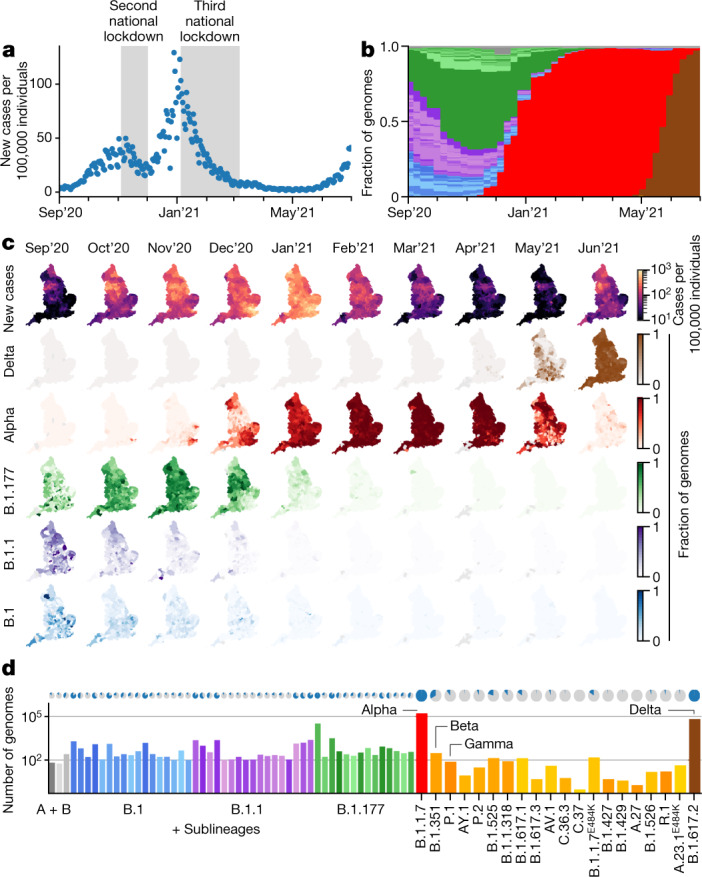
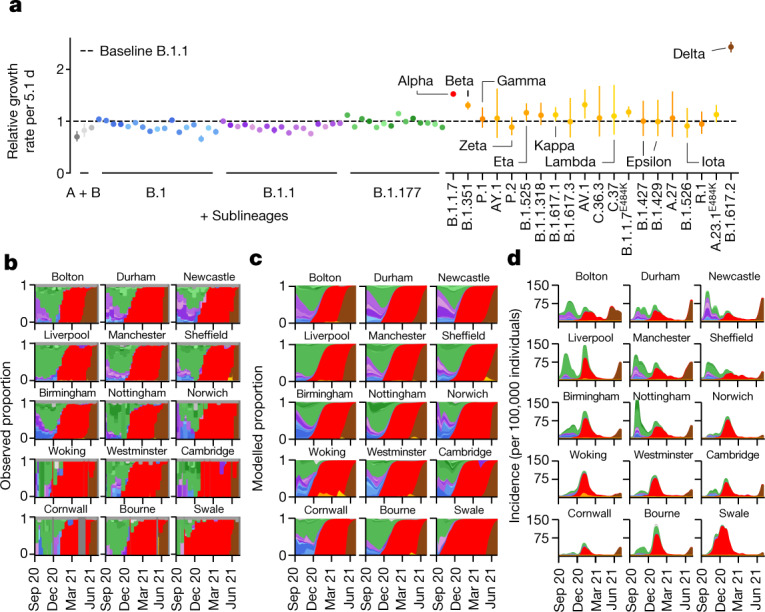
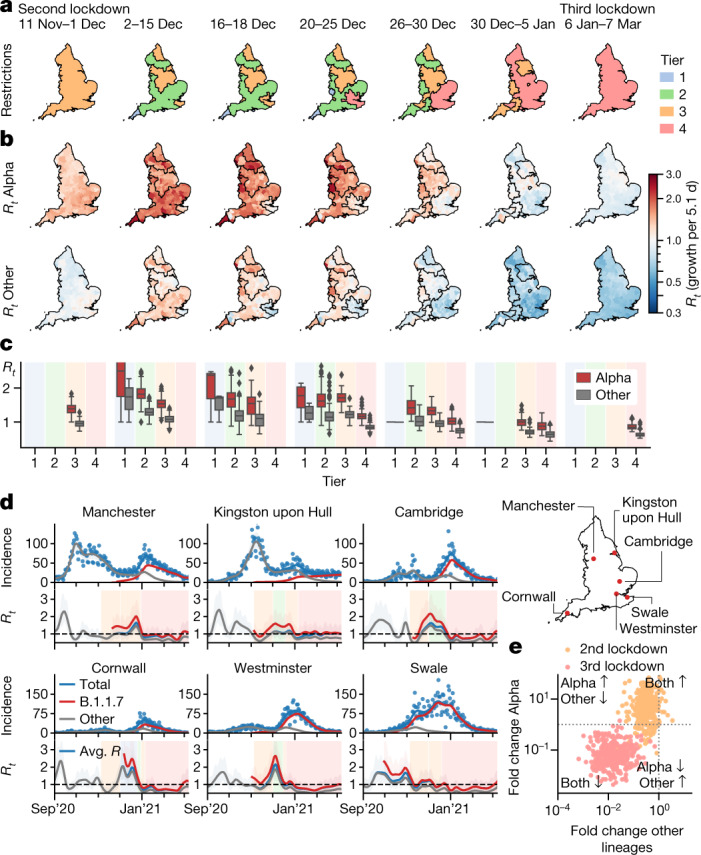
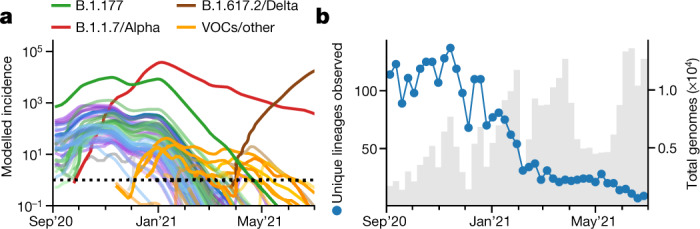
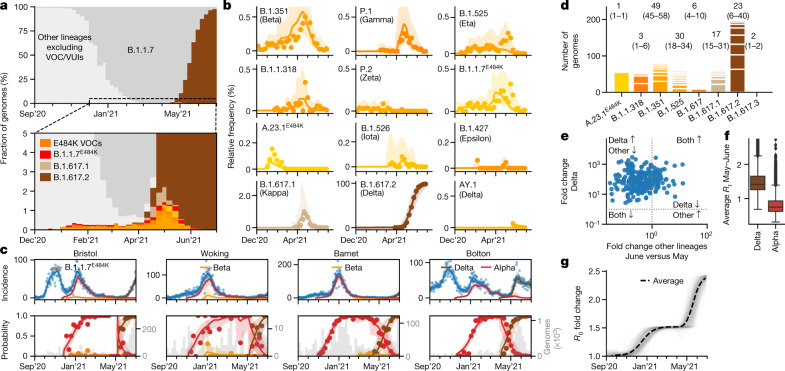
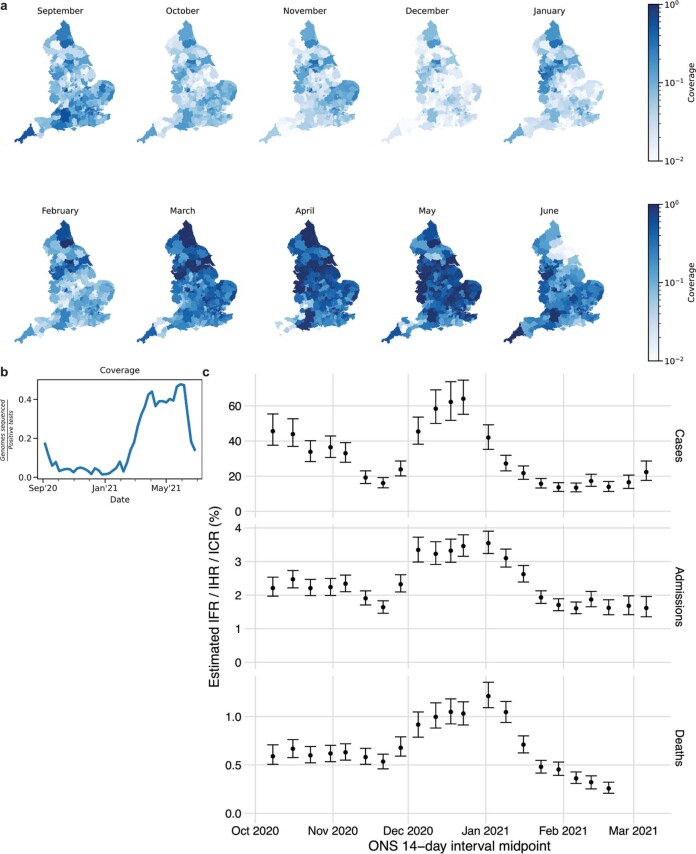
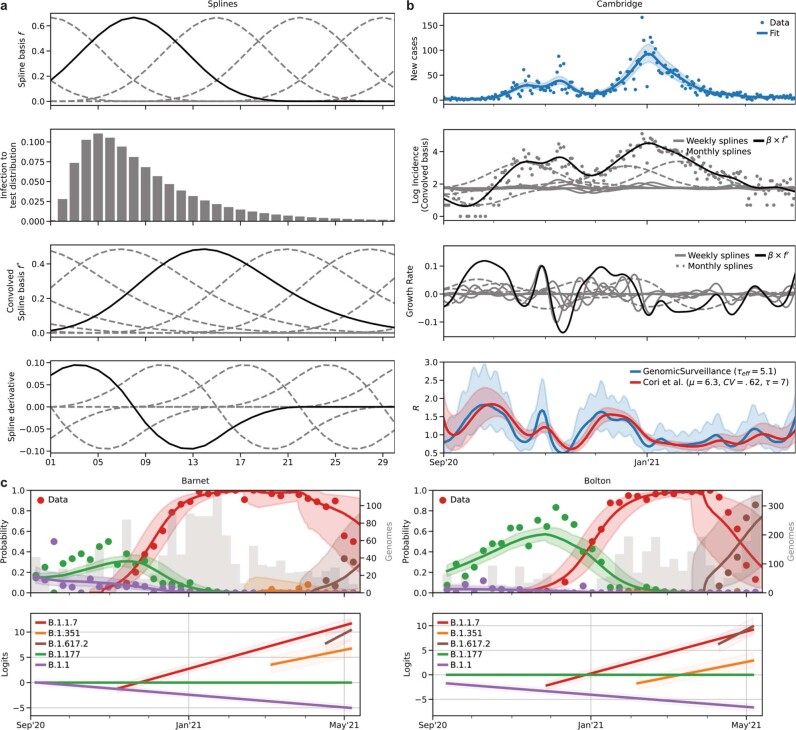
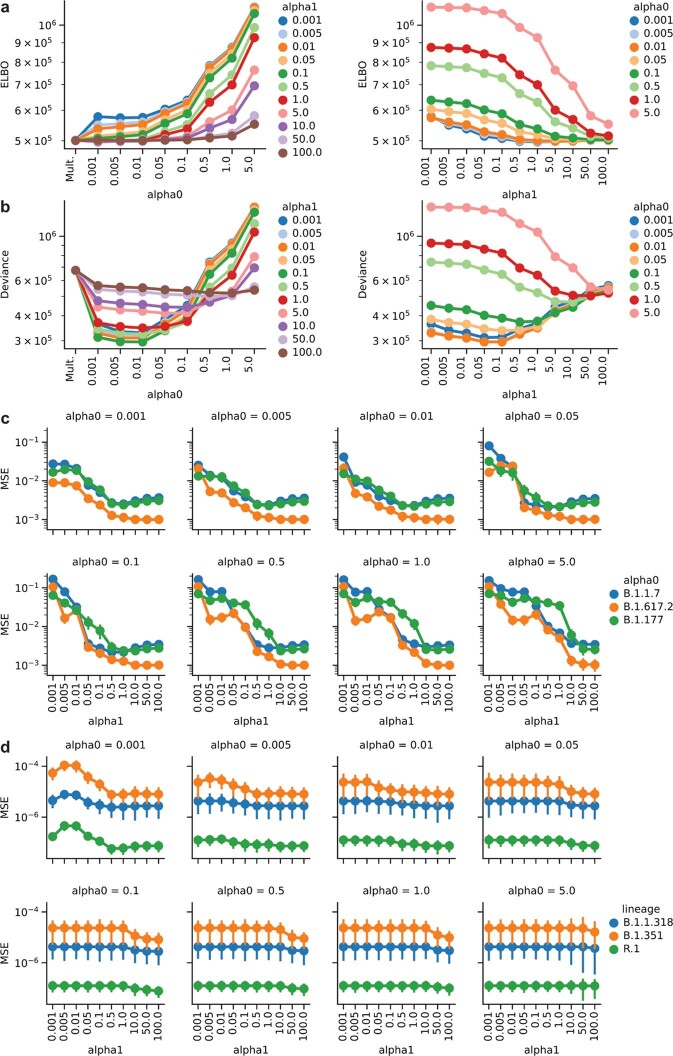
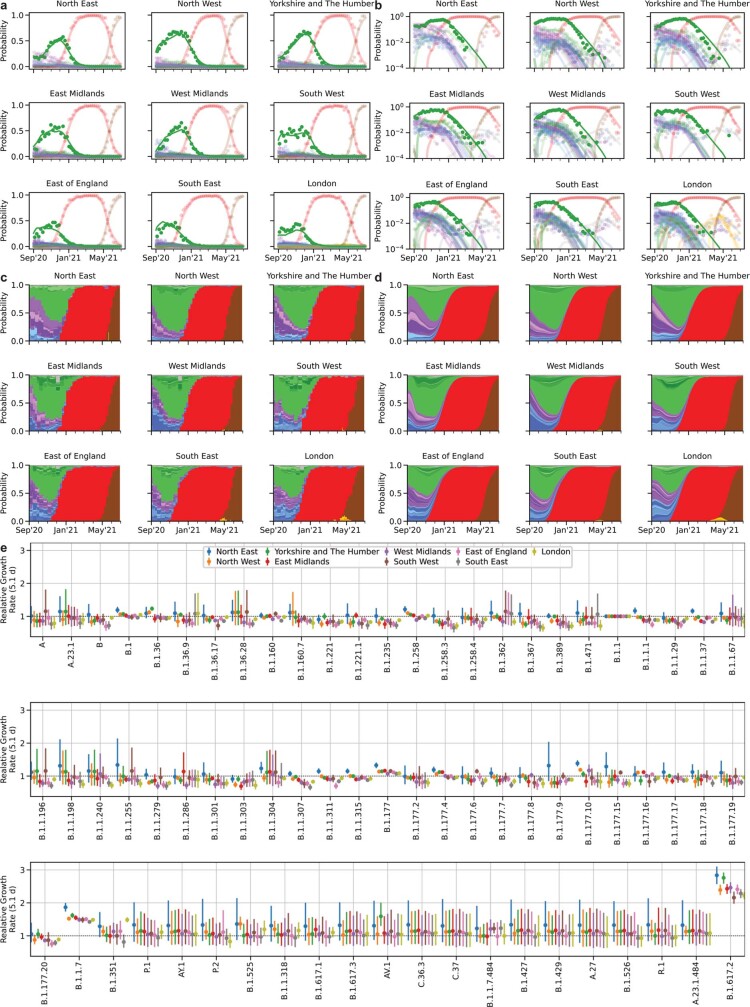
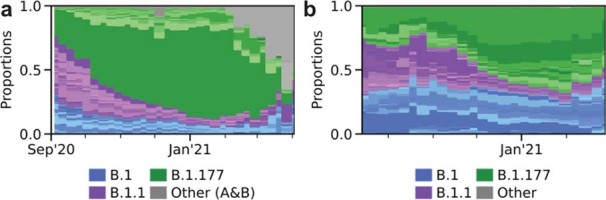
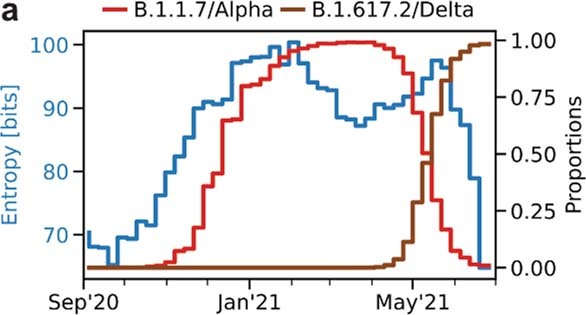
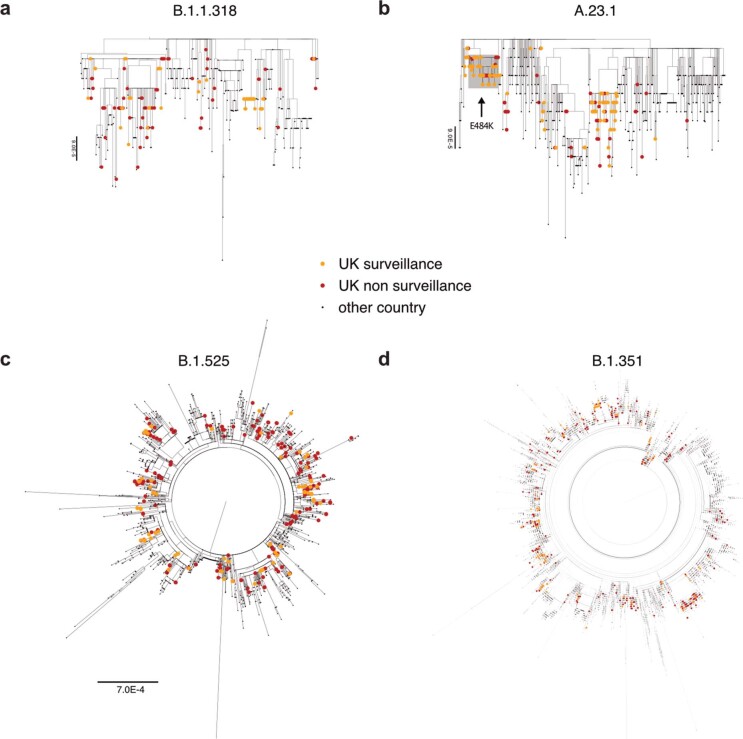
The evolution of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus leads to new variants that warrant timely epidemiological characterization. Here we use the dense genomic surveillance data generated by the COVID-19 Genomics UK Consortium to reconstruct the dynamics of 71 different lineages in each of 315 English local authorities between September 2020 and June 2021. This analysis reveals a series of subepidemics that peaked in early autumn 2020, followed by a jump in transmissibility of the B.1.1.7/Alpha lineage. The Alpha variant grew when other lineages declined during the second national lockdown and regionally tiered restrictions between November and December 2020. A third more stringent national lockdown suppressed the Alpha variant and eliminated nearly all other lineages in early 2021. Yet a series of variants (most of which contained the spike E484K mutation) defied these trends and persisted at moderately increasing proportions. However, by accounting for sustained introductions, we found that the transmissibility of these variants is unlikely to have exceeded the transmissibility of the Alpha variant. Finally, B.1.617.2/Delta was repeatedly introduced in England and grew rapidly in early summer 2021, constituting approximately 98% of sampled SARS-CoV-2 genomes on 26 June 2021.
© 2021. The Author(s).
Conflict of interest statement
E.B. is a paid consultant of Oxford Nanopore.
Figures
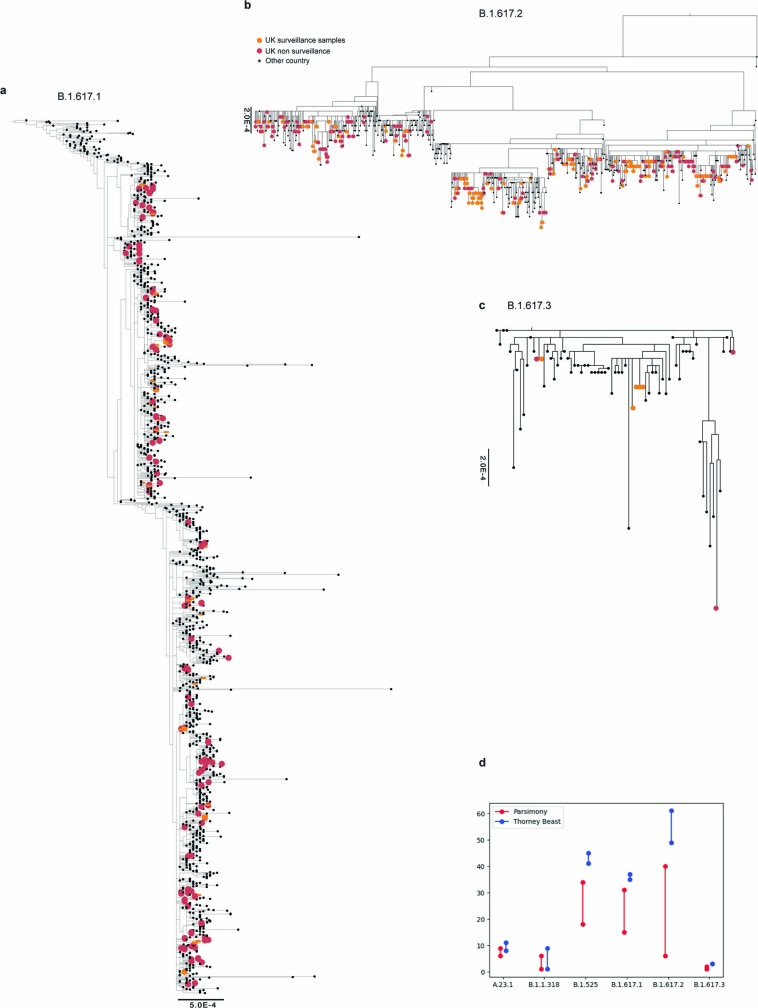
References
-
- Rambaut, A. Phylogenetic Analysis of nCoV-2019 Genomes (Virological, 2020); https://virological.org/t/phylodynamic-analysis-176-genomes-6-mar-2020/356
-
- Nextstrain Team Genomic Epidemiology of Novel Coronavirus—Global Subsampling (Nextstrain, 2020); https://nextstrain.org/ncov/global?l=clock
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- MC_PC_19012/MRC_/Medical Research Council/United Kingdom
- 21777/CRUK_/Cancer Research UK/United Kingdom
- 210758/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- MC_UU_12014/12/MRC_/Medical Research Council/United Kingdom
- MR/R015600/1/MRC_/Medical Research Council/United Kingdom
- 204911/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_19026/MRC_/Medical Research Council/United Kingdom
- 220885/Z/20/Z/WT_/Wellcome Trust/United Kingdom
- 210818/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- 210918/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_19027/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
