

In-depth phenotyping for clinical stratification of Gaucher disease
- PMID: 34649574
- PMCID: PMC8515714
- DOI: 10.1186/s13023-021-02034-6
In-depth phenotyping for clinical stratification of Gaucher disease
Abstract
Background: The Gaucher Investigative Therapy Evaluation is a national clinical cohort of 250 patients aged 5-87 years with Gaucher disease in the United Kingdom-an ultra-rare genetic disorder. To inform clinical decision-making and improve pathophysiological understanding, we characterized the course of Gaucher disease and explored the influence of costly innovative medication and other interventions. Retrospective and prospective clinical, laboratory and radiological information including molecular analysis of the GBA1 gene and comprising > 2500 variables were collected systematically into a relational database with banking of collated biological samples in a central bioresource. Data for deep phenotyping and life-quality evaluation, including skeletal, visceral, haematological and neurological manifestations were recorded for a median of 17.3 years; the skeletal and neurological manifestations are the main focus of this study.
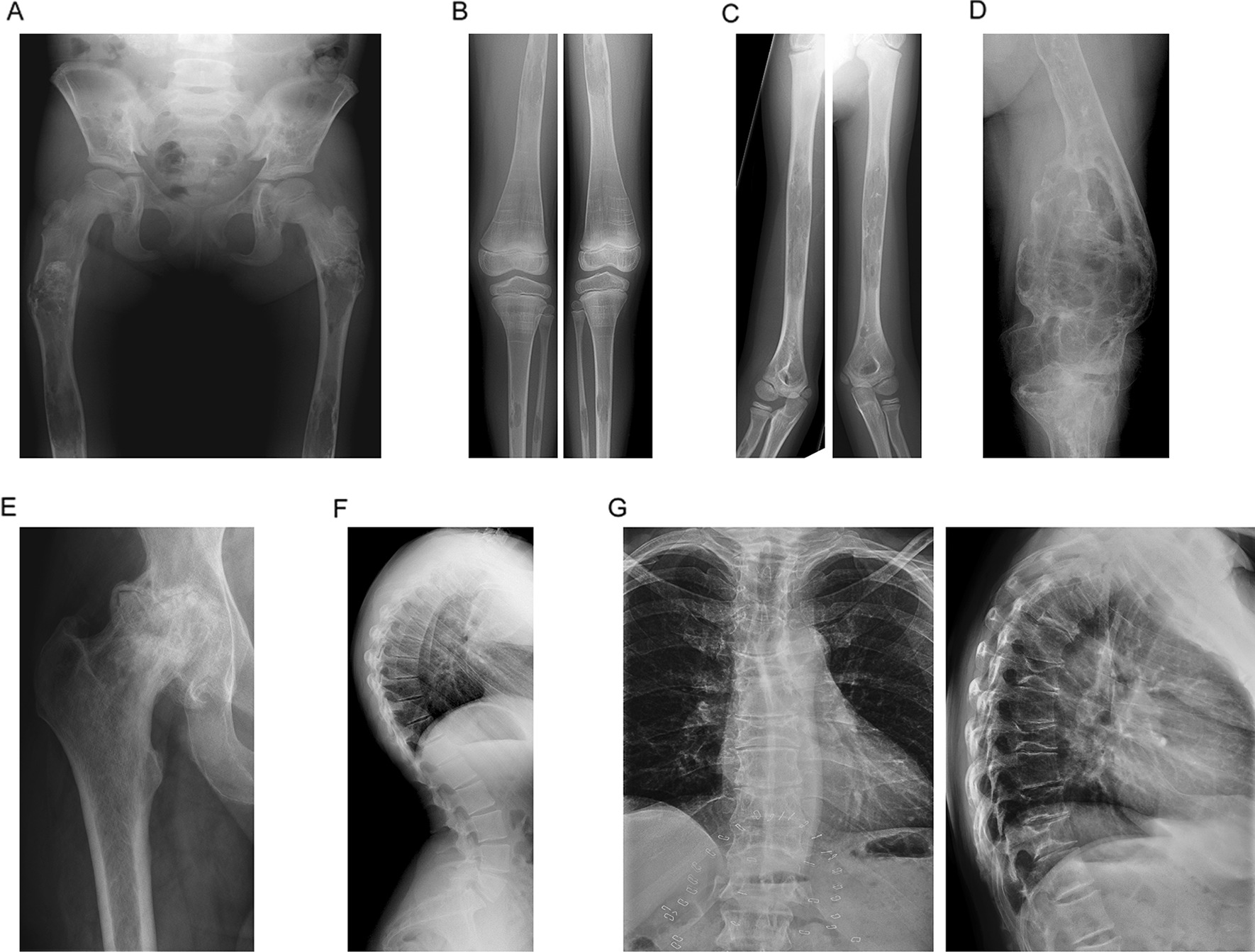
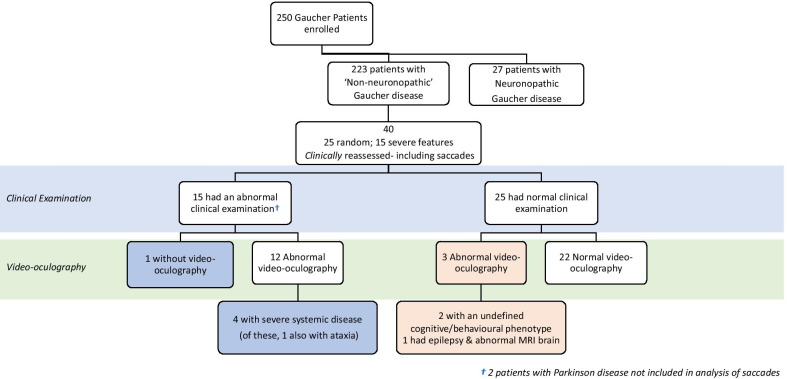
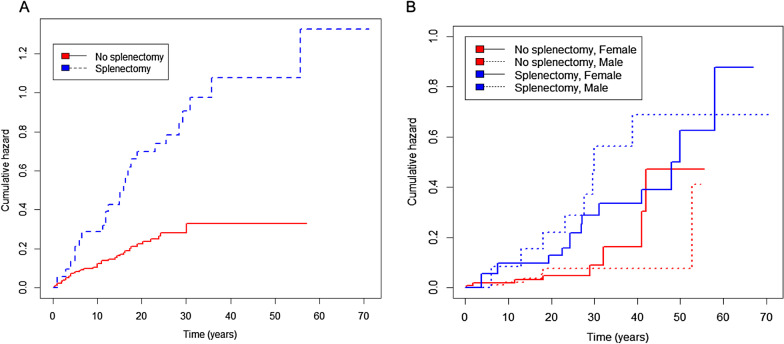
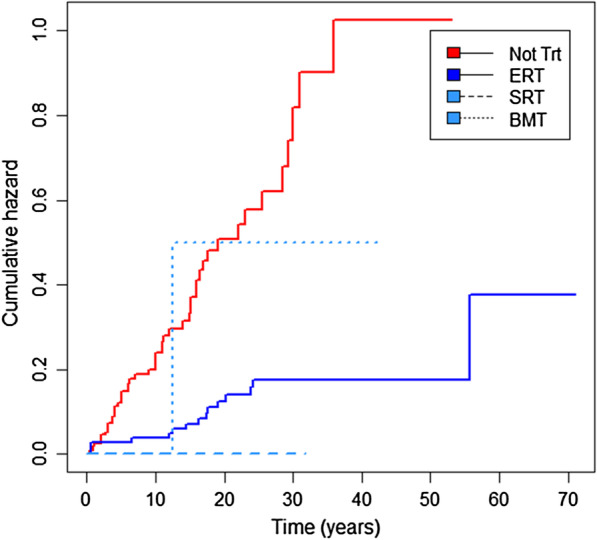
Results: At baseline, 223 of the 250 patients were classified as type 1 Gaucher disease. Skeletal manifestations occurred in most patients in the cohort (131 of 201 specifically reported bone pain). Symptomatic osteonecrosis and fragility fractures occurred respectively in 76 and 37 of all 250 patients and the first osseous events occurred significantly earlier in those with neuronopathic disease. Intensive phenotyping in a subgroup of 40 patients originally considered to have only systemic features, revealed neurological involvement in 18: two had Parkinson disease and 16 had clinical signs compatible with neuronopathic Gaucher disease-indicating a greater than expected prevalence of neurological features. Analysis of longitudinal real-world data enabled Gaucher disease to be stratified with respect to advanced therapies and splenectomy. Splenectomy was associated with an increased hazard of fragility fractures, in addition to osteonecrosis and orthopaedic surgery; there were marked gender differences in fracture risk over time since splenectomy. Skeletal disease was a heavy burden of illness, especially where access to specific therapy was delayed and in patients requiring orthopaedic surgery.
Conclusion: Gaucher disease has been explored using real-world data obtained in an era of therapeutic transformation. Introduction of advanced therapies and repeated longitudinal measures enabled this heterogeneous condition to be stratified into obvious clinical endotypes. The study reveals diverse and changing phenotypic manifestations with systemic, skeletal and neurological disease as inter-related sources of disability.
Keywords: Cohort; Disease-modifying therapies; Enzyme replacement therapy; GAUCHERITE; Gaucher disease; Substrate reduction therapy.
© 2021. The Author(s).
Conflict of interest statement
SHIRE Biopharmaceuticals UK (now part of Takeda) supported the licence fee for the SF-36v2 questionnaire (< $6000). The authors confirm that neither SHIRE nor any other external agency played any part in the study design, data collection, analysis—nor interpretation. The named authors are solely responsible for the manuscript and the decision to submit it for publication.
Figures
References
-
- Beutler E, Grabowski GA. The metabolic and molecular basis of inherited disease. 8. New York: McGraw-Hill Book Co.; 2001.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
