

High-confidence structural annotation of metabolites absent from spectral libraries
- PMID: 34650271
- PMCID: PMC8926923
- DOI: 10.1038/s41587-021-01045-9
High-confidence structural annotation of metabolites absent from spectral libraries
Abstract
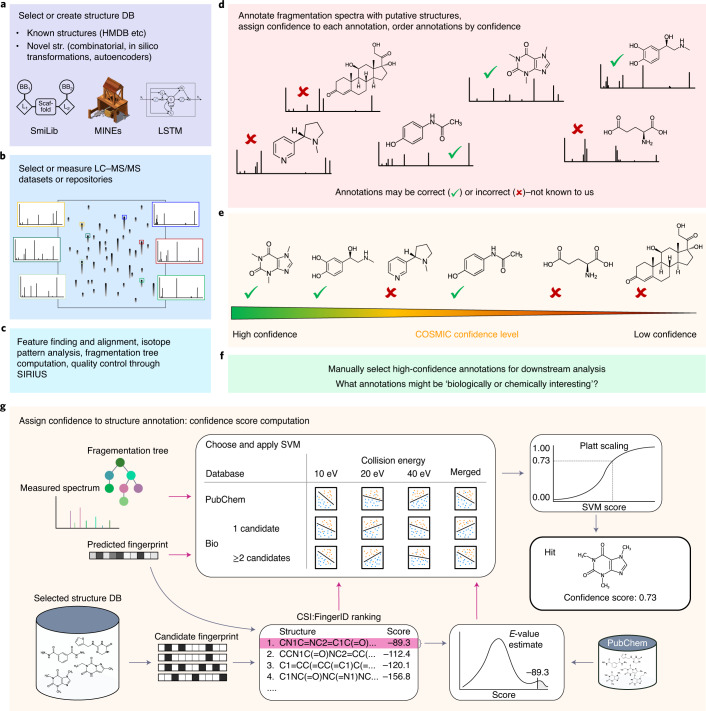
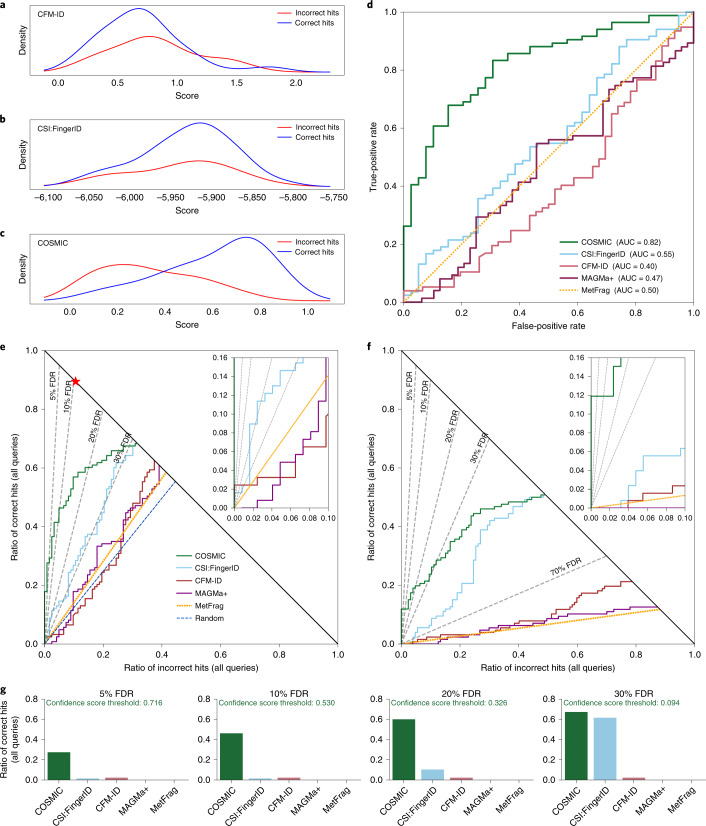
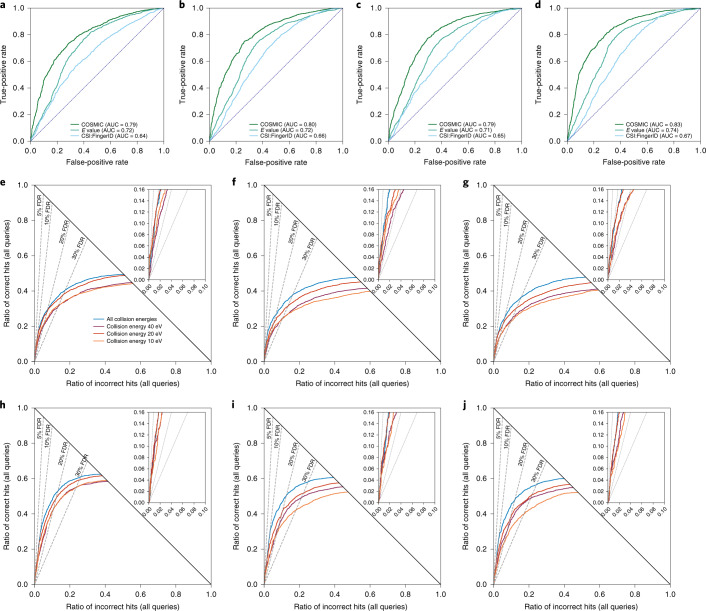
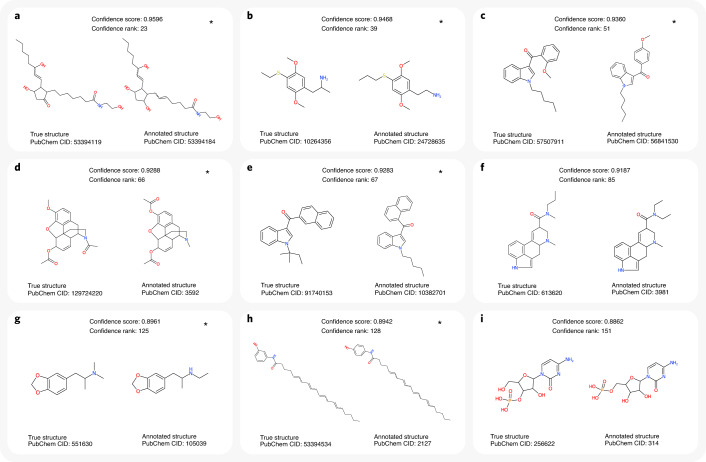
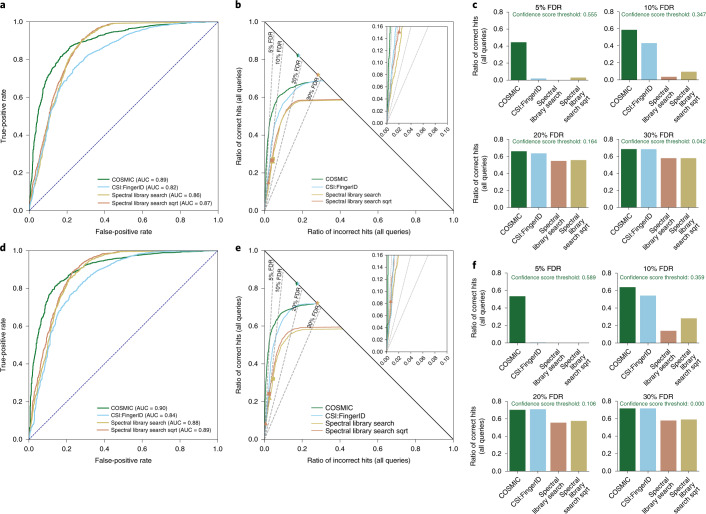
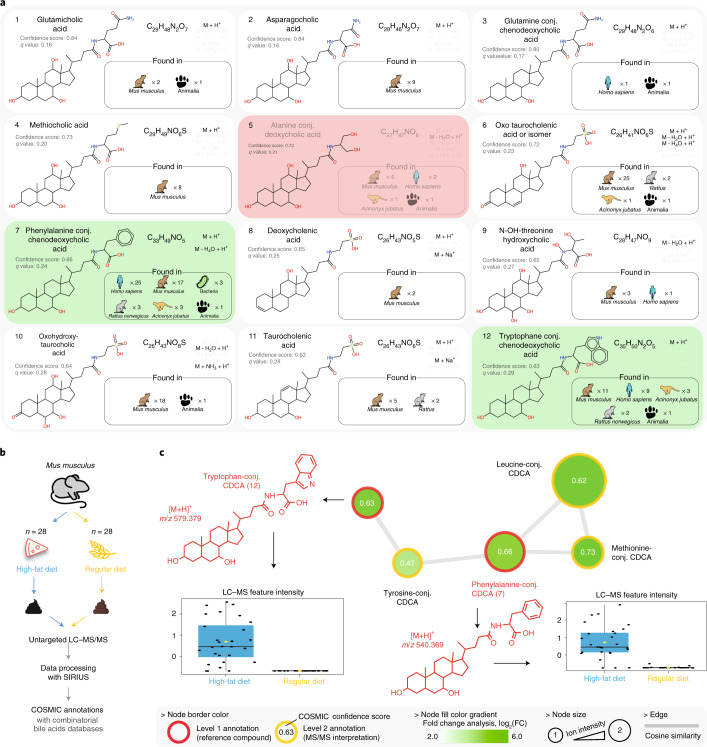
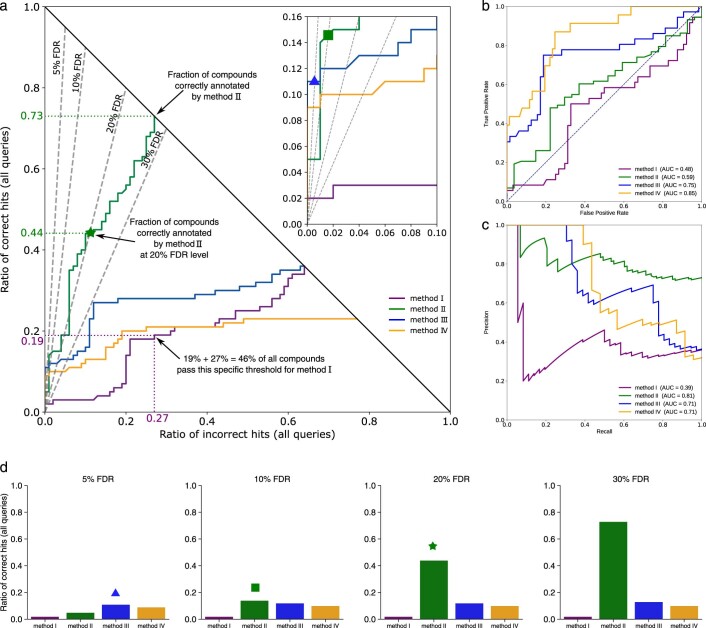
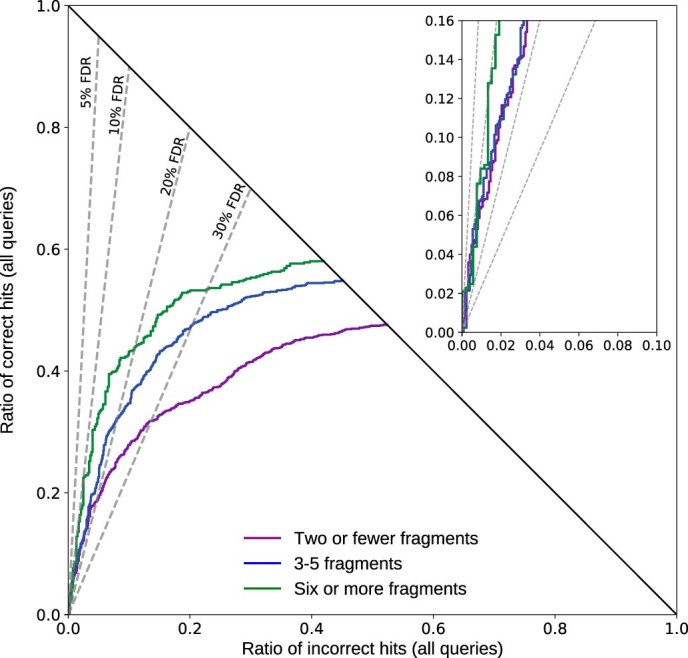
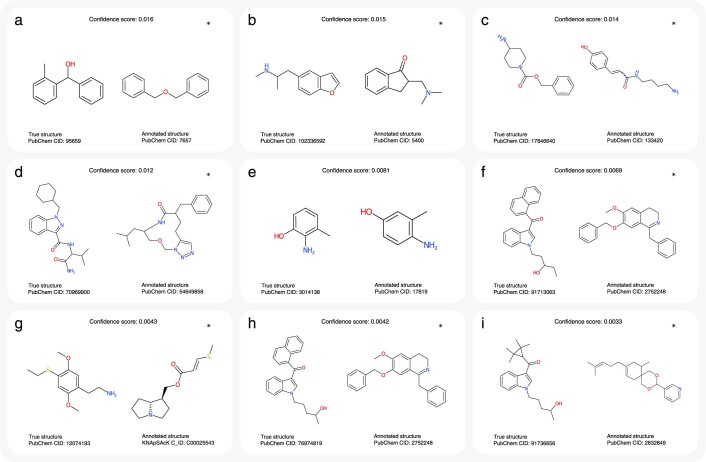
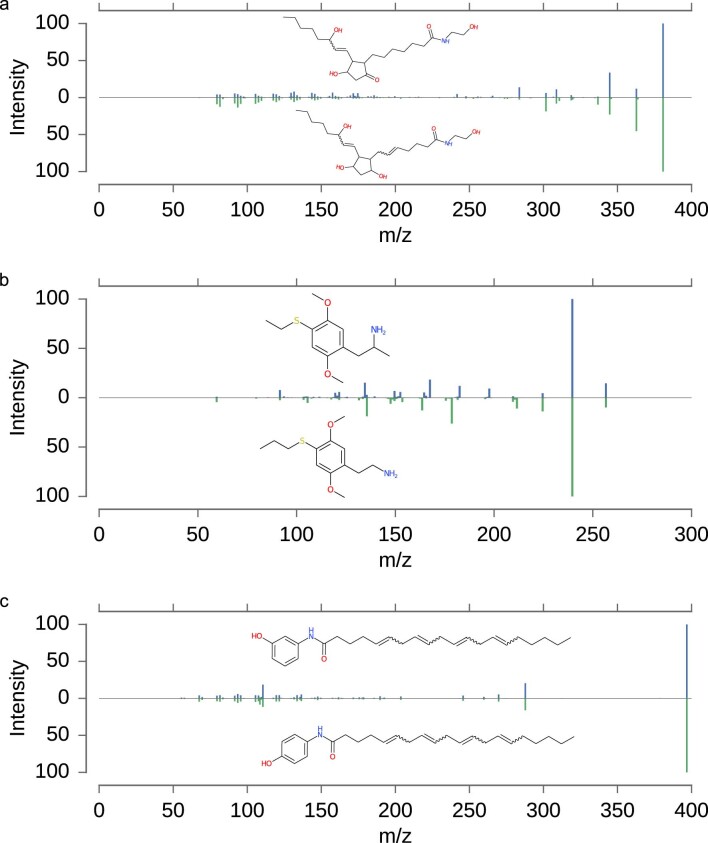
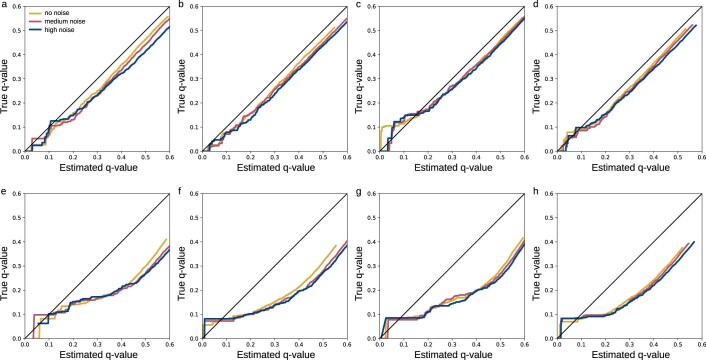
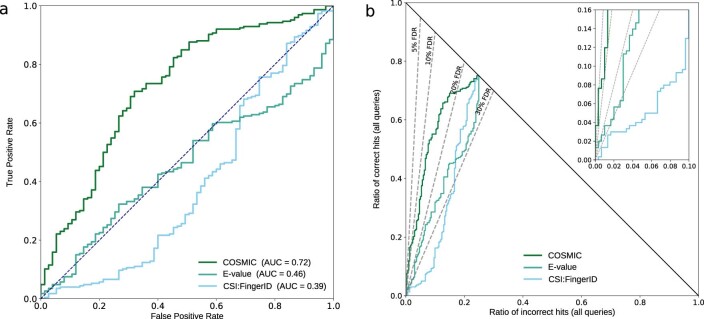
Untargeted metabolomics experiments rely on spectral libraries for structure annotation, but, typically, only a small fraction of spectra can be matched. Previous in silico methods search in structure databases but cannot distinguish between correct and incorrect annotations. Here we introduce the COSMIC workflow that combines in silico structure database generation and annotation with a confidence score consisting of kernel density P value estimation and a support vector machine with enforced directionality of features. On diverse datasets, COSMIC annotates a substantial number of hits at low false discovery rates and outperforms spectral library search. To demonstrate that COSMIC can annotate structures never reported before, we annotated 12 natural bile acids. The annotation of nine structures was confirmed by manual evaluation and two structures using synthetic standards. In human samples, we annotated and manually validated 315 molecular structures currently absent from the Human Metabolome Database. Application of COSMIC to data from 17,400 metabolomics experiments led to 1,715 high-confidence structural annotations that were absent from spectral libraries.
© 2022. The Author(s).
Conflict of interest statement
S.B., K.D., M.L., M.F. and M.A.H. are co-founders of Bright Giant. P.C.D. is scientific advisor for Sirenas, Galileo and Cybele and is scientific advisor and co-founder of Enveda and Ometa. The remaining authors declare no competing interests.
Figures
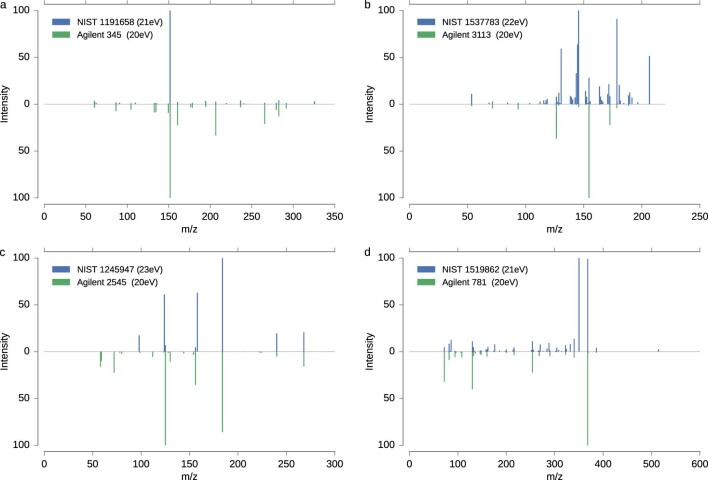
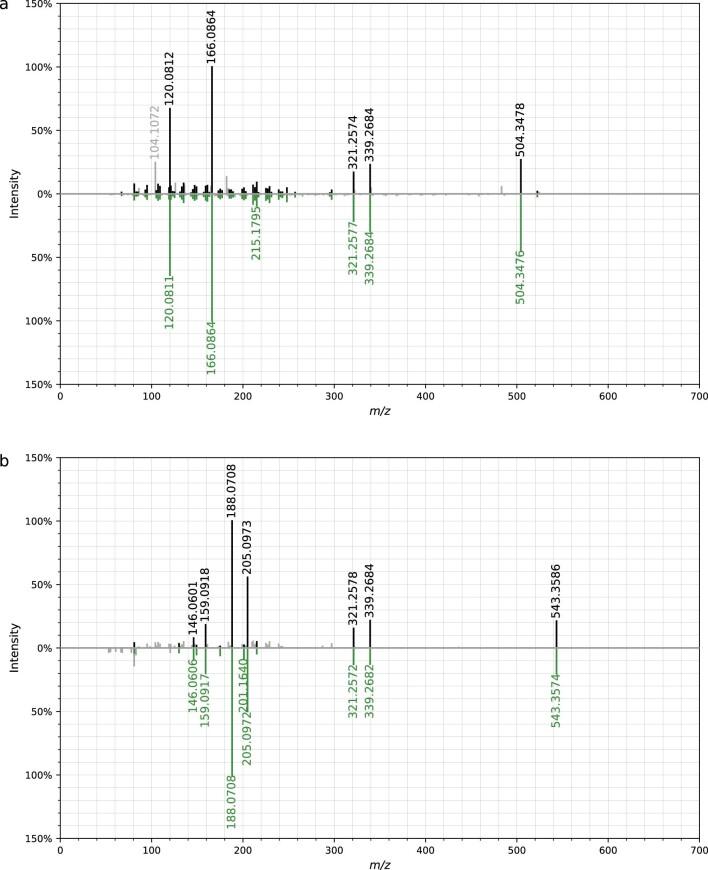
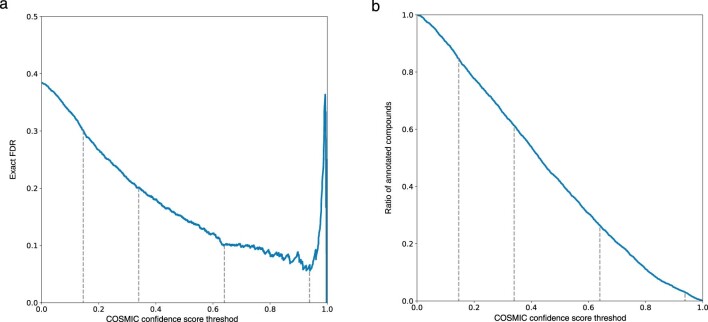

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
